
This section is an adaptation of the paper: “DICER1: mutations, microRNAs and mechanisms”, published in Nature Reviews Cancer by Foulkes, Priest and Duchaine (PMID 25176334). For more information, please refer to the original article.
For an up to date list of papers published about the DICER1 gene and DICER1 syndrome, please click here.
Molecular Functions
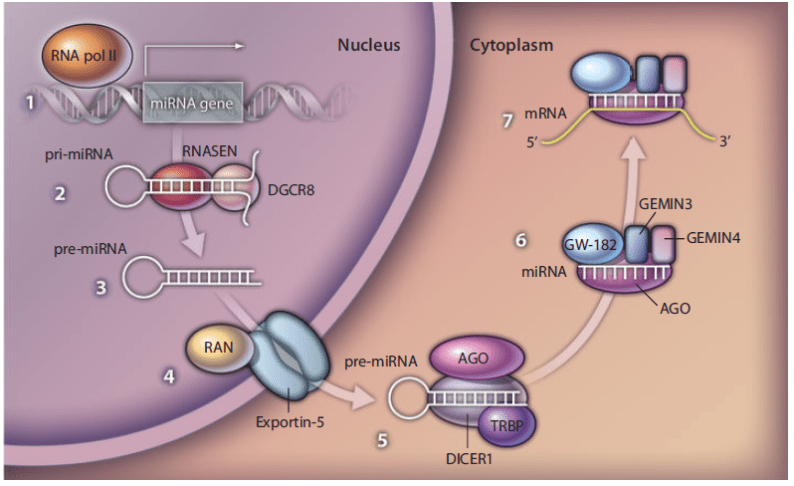
The Dicer protein is an endoribonuclease responsible for the production of mature microRNAs (miRNAs), which are small single-stranded RNA molecules that target messenger RNAs (mRNAs) and have an important role in regulating gene expression by repressing protein synthesis. The human form of the Dicer protein is called DICER1, encoded by the DICER1 gene, and is widely expressed across tissues. DICER1 is involved in the miRNA-mediated gene silencing pathway, where a miRNA imperfectly hybridizes to target mRNAs. This, in turn, generally leads to repression of mRNA translation and initiation of mRNA decay. A primary miRNA (pri-miRNA) transcript is produced via transcription of a miRNA gene by the RNA polymerase II enzyme. The pri-miRNA is then processed in the nucleus by DROSHA (also known as RNASEN), an RNA endonuclease, and the RNA binding protein DGCR8 (also known as PASHA), resulting in the production of an RNA stemloop that contains the miRNA. This premature miRNA (pre-miRNA) hairpin is subsequently exported into the cytoplasm through nuclear pores. The cytoplasmic DICER1 protein then cleaves the pre-miRNA to release the mature short (~21 nucleotide) miRNA. This miRNA is then loaded into an Argonaute (AGO) protein forming the core of the miRNA-induced silencing complex (miRISC). The miRISC then hybridizes to targeted mRNAs in order to engender their silencing.
DICER1 Structure
- The human DICER1 gene is located on chromosome 14q32.13 and is comprised of 27 exons with 1,922 amino acids. The DICER1 protein is a large multi-domain enzyme shaped like an ‘L’.
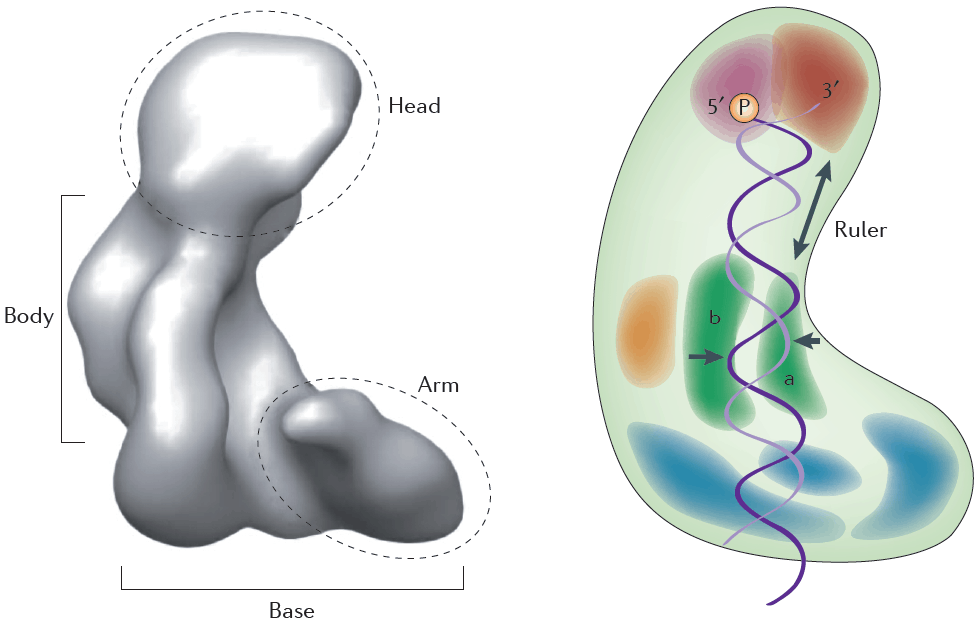
- At the top of the ‘L’ are the PAZ and Platform domains. These domains are binding pockets for double-stranded RNA (dsRNA). The 3’-overhang of the dsRNA substrate binds at the PAZ domain, and the 5’-overhang binds at the Platform domain.
- The lower half of DICER1 consists of the RNase IIIa and RNase IIIb domains, which dimerize to form the critical catalytic core of the enzyme and each cleave one strand of the dsRNA substrate. The RNase IIIa domain is responsible for producing 3p miRNAs from the 3’ strand, and the RNase IIIb domain produces 5p miRNAs from the 5’ strand. DICER1 requires magnesium and/or manganese ions in order to perform the catalytic cleavage. These metal ions are bound to ion-binding residues within the RNase III domains at amino acid residues E1320 and E1564 in the RNase IIIa domain, and D1709 and E1813 in the RNase IIIb domain.
- Clinical correlation: Most individuals with DICER1 syndrome-related tumors are found to harbour two mutations in DICER1. These typically consist of one heterozygous germline truncating (inactivating) mutation, which may be located at any position throughout the gene, coupled with a somatic (or tumor-specific) missense mutation, which consistently affect one of the metal-ion binding resides within the RNase IIIb domain or an adjacent residue. Almost all tumors and dysplasias associated with the syndrome have been found to harbour such ‘hotspot’ mutations. In a very small subgroup of patients with DICER1 syndrome, the initial mutational events are mosaic RNase IIIb mutations; the phenotype in these cases may be more severe
- The RNase III domains are separated from the dsRNA binding domains by a linker, which contains a connector helix. This holds the dsRNA phosphate backbone in place. It also acts as a ‘ruler’ by positioning the pre-miRNA along the enzyme so that the enzyme can cleave the dsRNA into miRNAs of appropriate sizes.
- At the base of the L is the DExD/H box helicase domain. This domain is thought to clamp down on the dsRNA substrate.
Tumor Spectrum
