Síndrome de DICER1: genética, herencia y características clínicas clave

El síndrome de DICER1 es un síndrome de predisposición tumoral familiar que se genera a partir de mutaciones genéticas en un gen denominado DICER1. Algunos tumores del síndrome de DICER1 son muy graves y mortales, pero muchos no son tan graves. Por lo general, los tumores son raros o muy raros, y algunos son tan raros que pueden ocurrir solo en personas con una mutación de DICER1.
Al parecer, no existe ningún patrón para el desarrollo de los tumores, excepto que cada tumor del síndrome de DICER1 tiende a producirse en ciertas edades típicas para ese tumor. Aquellos que desarrollan los diferentes tumores asociados con el síndrome de DICER1 tienden a hacerlo en la niñez, incluso en la infancia, o alrededor de los 30 años. Se estima que más de la mitad de las personas susceptibles al síndrome de DICER1 no tienen ningún problema médico.
¿Que es el gen DICER1 y cual es su función?
En pocas palabras, el gen DICER1 es una instrucción para cada célula humana, que le indica a la célula qué otra instrucción genética debe escuchar. Más específicamente, el gen DICER1 le indica a la célula que genere una proteína que sea un controlador importante de las actividades celulares. Casi todos los organismos vivos requieren este tipo de control, por lo que la mayoría de los organismos vivos tiene genes dicer. “DICER1” es el nombre específico de la versión humana del gen dicer que, en realidad, se descubrió en primer lugar en un pequeño gusano.
Patrón de herencia
Seres humanos tienen dos copias de gen DICER1 en cada célula. Por lo general, la susceptibilidad al síndrome de DICER1 es el resultado de heredar una copia anormal (“mutada”) del gen DICER1. Como las células tienen dos copias de cada gen, habitualmente las células funcionan satisfactoriamente con una copia anormal. La herencia de un gen mutado es del padre o de la madre del paciente. Científicamente, el patrón de herencia del síndrome de DICER1 se llama herencia “autosómica dominante”. Un padre tiene una copia normal y una copia mutada del gen DICER1. Un hijo de este padre heredará la copia normal o la copia anormal. Las posibilidades de la versión a heredar son del 50 %. Cada hijo de este padre tiene una posibilidad del 50 %. Las posibilidades son que la mitad de los hijos del padre hereden la copia normal y la otra mitad herede la copia mutada, y solamente estos niños serán susceptibles a desarrollar enfermedades del síndrome de DICER1. Tal mutación está presente en todas las células del cuerpo de una persona y se puede detectar en el cabello, la saliva, la sangre, etcétera. Se estima que las mutaciones de DICER1 solo aparecen en 1 de cada 10,000 bebés al nacer; por lo tanto, sería extremadamente raro que dos padres tengan una mutación de DICER1, nunca se ha observado un niño con mutaciones de DICER1 de ambos padres.
En alrededor del 10 al 15 % de los casos, la mutación de DICER1 no se hereda de un padre. En cambio, un niño tiene una nueva mutación; no se conoce si la mutación se produjo en una célula del esperma, en el óvulo de la madre, o muy al inicio después de la concepción. A partir de ese entonces, una persona con una nueva mutación puede transmitir la mutación a sus hijos.
Existen muy pocas personas en las que la mutación del DICER1 está presente solo en algunos tejidos de su cuerpo, por ejemplo, en las células del pulmón o en células del riñón, o en cualquier otro tejido específico. Aún se desconoce cómo se producen tales mutaciones o por qué se distribuyen solamente a ciertos tejidos. Sólo los tejidos con la mutación son susceptibles a enfermedades asociadas con DICER1. Estas mutaciones se denominan “mosaico” porque solo aparecen en algunas de las células del cuerpo.
¿Cuales son las señales que una persona tiene una mutacion DICER1?
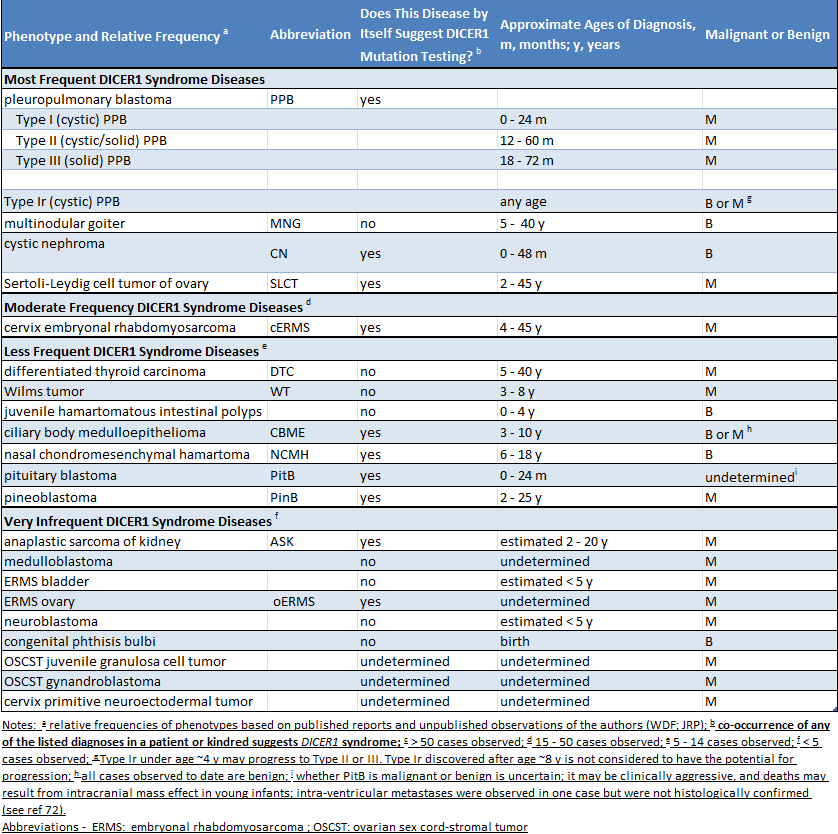
Como muchas personas con una mutación de DICER1 no tienen problemas médicos, es muy posible que el padre no sea consciente de que tienen una mutación. La sospecha de una mutación se produce por cualquiera de dos circunstancias. En primer lugar, si un niño es diagnosticado con una enfermedad estrechamente relacionada con el síndrome de DICER1 (como blastoma pleuropulmonar), se debe investigar la mutación de DICER1. En segundo lugar, si se diagnostica alguna de las dos o más enfermedades relacionadas con el síndrome de DICER1 en un niño o en sus hermanos u otros parientes cercanos, es razonable que se investigue la mutación de DICER1. La siguiente tabla enumera las enfermedades del síndrome de DICER1. La tabla indica si las enfermedades enumeradas están principalmente asociadas con el síndrome de DICER1 (columna 3, “sí”) o si ocurren en la población en general, y también más raramente en el síndrome de DICER 1 (columna 3, “no”).
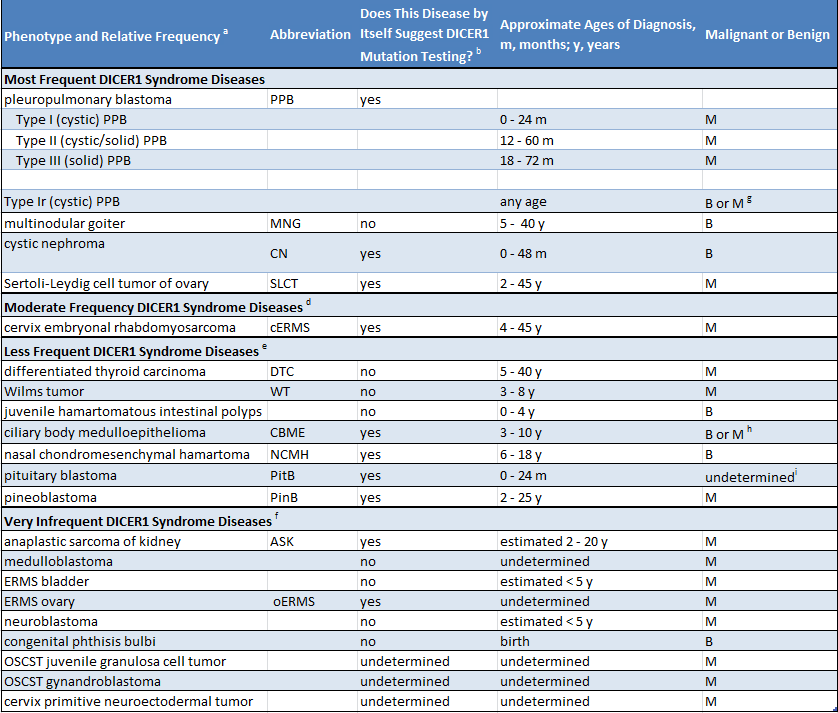
(Click to enlarge)
A continuación se presentan ejemplos de cuándo se debe sospechar del síndrome de DICER1. Si se diagnostica a un niño con blastoma pleuropulmonar (PPB) o blastoma pituitario, la sospecha de presentar una mutación de DICER1 es alta, ya que aproximadamente el 75 % de los pacientes con PPB tienen una mutación de DICER1 y más del 90 % de los pacientes con blastoma pituitario tienen una mutación. En cambio, si se diagnostica a un niño con tumor de Wilms en el riñón, la sospecha no es alta, ya que solo un porcentaje muy pequeño de los pacientes con tumor de Wilms (muy por debajo del 5 %) tienen una mutación de DICER1. Ciertas combinaciones de enfermedades (en una persona o en familiares cercanos) levantan excesivamente la sospecha de síndrome de DICER1: por ejemplo, cerca del 50 % de los tumores ováricos de células de Sertoli-Leydig son causados por mutaciones de DICER1, pero cuando este tumor ocurre con nódulos en la tiroides (bocio multinodular) en el paciente o en familiares, la combinación sugiere firmemente que una mutación de DICER1 está causando ambas enfermedades.
Mutaciones DICER1 en tumores
Por último, esta discusión sobre la genética del DICER1 debe incluir un hecho acerca de los genes DICER1 en los tumores mismos. Los científicos han determinado que los tumores suelen tener mutaciones en ambas copias del gen DICER1. La primera mutación, en una copia del gen, es la “mutación constitucional” de la persona (presente en todas las células). La segunda mutación afecta a la segunda copia del gen y solo está presente en las células tumorales. Aún se desconoce cómo, cuándo y por qué se produce la segunda mutación. Sin embargo, se cree que el tumor se desarrolla porque ninguna copia del gen era normal. Incluso, las enfermedades benignas asociadas con el DICER1 tienen dos copias mutadas del gen.
Pleuropulmonary Blastoma

El blastoma pleuropulmonar (PPB) es un tumor de pulmón raro y maligno que aparece en la primera infancia. Fue reconocido por los médicos, en primer lugar, alrededor de 1985. Por supuesto, el PPB ocurrió antes de 1985, pero era tan raro que los médicos nunca los habían reconocido como una enfermedad distinta. El PPB es totalmente distinto y no tiene ninguna relación con el cáncer de pulmón en los adultos. Debido a que esta rara enfermedad, PPB, ocurría en algunas familias, la investigación de la causa condujo al descubrimiento de las mutaciones de DICER1, que ahora son conocidas por causar la mayoría de los casos de PPB.
¿Cuando ocurre?
El PPB se produce casi exclusivamente en niños menores de 6 años. Puede ocurrir en bebés. La mayoría de los niños con PPB tiene una mutación del DICER1 y pocas veces ocurren casos sin tal mutación. El PPB ocurre muy raramente en niños mayores y adultos.
¿Que tipos de PPB existen?
El PPB ocurre en tres formas básicas: en primer lugar, en niños menores de 1 año, el PPB ocurre en forma de “quistes” en el pulmón (los quistes del PPB son cavidades llenas de aire productos del crecimiento anormal del pulmón). Tales quistes pueden ser pequeños y no causar ningún síntoma, mientras que otros pueden ser muy grandes y causar dificultades respiratorias leves o severas. El PPB enquistado se denomina PPB Tipo I.
La mayoría de los quistes pulmonares en bebés NO son PPB; se trata de una enfermedad completamente diferente y menos grave, y no tiene nada que ver con las mutaciones de DICER1. La cirugía elimina los quistes sin problemas posteriores. Como el PPB es raro y otros quistes son más frecuentes, los médicos casi siempre consideran que los quistes de pulmón son el tipo más común. Sólo después de quitar y examinar un quiste con un microscopio se puede diagnosticar el PPB. Los rayos X se ven similares para los quistes de PPB y los quistes más comunes. Por lo general, la extirpación de quistes de PPB es curativa.
La segunda forma de PPB (PPB Tipo II) ocurre generalmente en niños de 12 a 30 meses. Estos PPB son una combinación de quistes y nódulos de tumores sólidos. La tercera forma de PPB es un tumor sólido sin quistes (PPB Tipo III) que ocurre generalmente en niños de 2 a 4 años. Los tumores de PPB de Tipo II y Tipo III pueden causar dificultad para respirar, o signos generales de una enfermedad o fiebre con una radiografía de pecho que muestra “sombras”. Como el PPB es raro en comparación con la neumonía, generalmente los médicos piensan inicialmente que un niño tiene neumonía. El PPB Tipo II y Tipo III son graves y malignos, y requieren cirugía y quimioterapia y, a veces, radioterapia. Muchos niños se curan de estos tumores, pero otros, lamentablemente, pueden morir debido a estas formas de PPB.
Como se señaló anteriormente, los rangos de edad del PPB Tipo I, II y III no son precisos. Además, el PPB Tipo I puede convertirse en PPB Tipo II o Tipo III, por lo tanto todos los tipos están conectados y representan un “espectro” de las manifestaciones del PPB. Hay una forma muy rara del PPB Tipo I llamada PPB Tipo Ir, que se cree que es una forma no maligna del PPB Tipo I, y no se convierte en el Tipo II ni en el Tipo III.
¿Cuando investigar una mutacion DICER1?
El PPB es muy característico de la mutación del DICER1. Por lo tanto, si un niño es diagnosticado con PPB, es razonable hacer una prueba para detectar mutaciones de DICER1.
Enfermedad tiroidea en el síndrome de DICER1
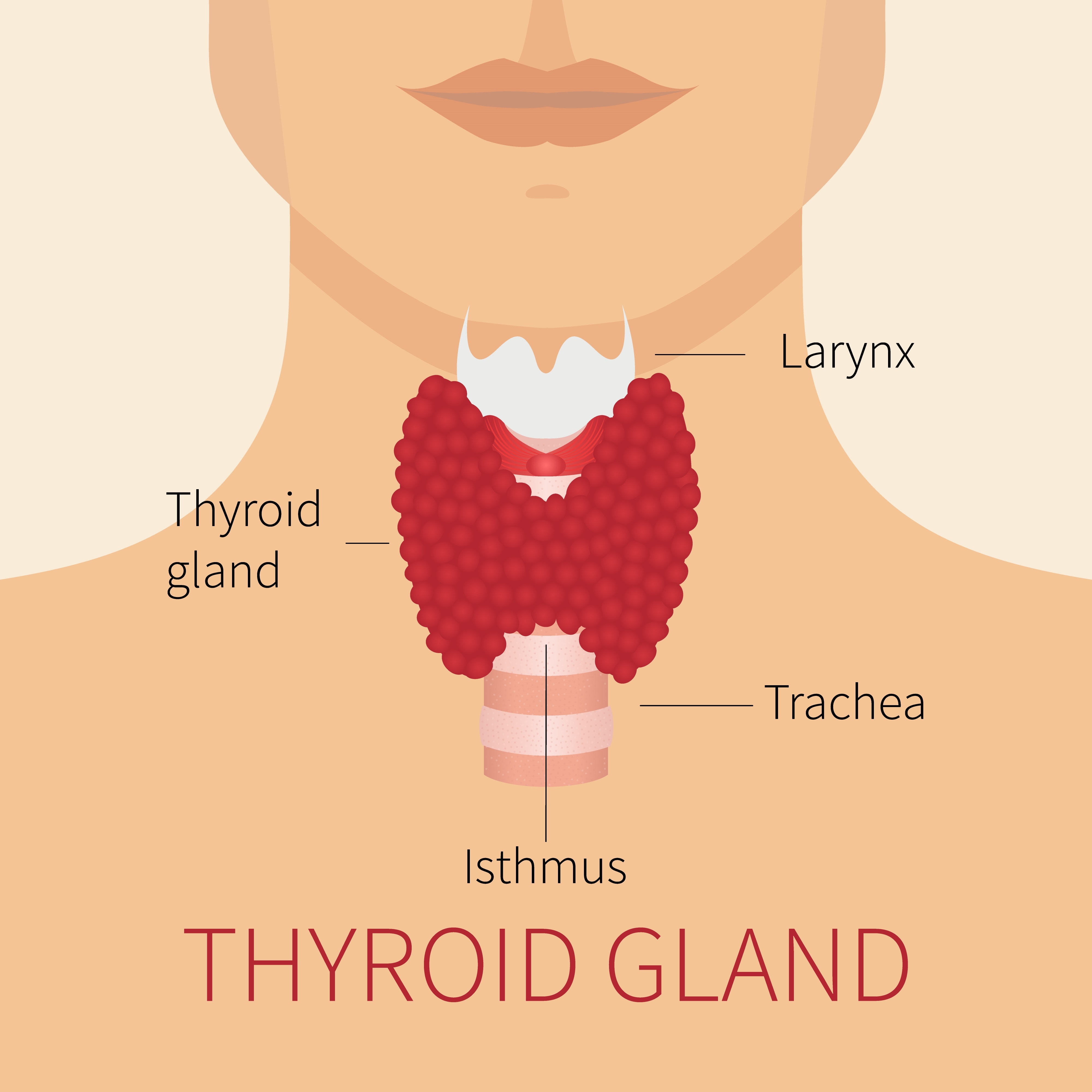
La glándula tiroides es el sitio de las anormalidades más frecuentes asociadas con las mutaciones de DICER1. La glándula se encuentra en la parte frontal del cuello, justo debajo de la laringe (“nuez de Adán”) y tiene lóbulos con forma de ala de mariposa a cada lado de la tráquea, como se muestra en el diagrama contiguo.
La glándula tiroides produce una hormona que controla muchas de las acciones del cuerpo que, generalmente, pueden describirse como ajuste del termostato de la energía del cuerpo. Cuando la hormona tiroidea es excesiva, el cuerpo adelgaza demasiado rápido y tiene una tendencia a sentirse demasiado caliente; cuando la hormona tiroidea es insuficiente, el cuerpo siente cansancio, frío, aumento de peso y estreñimiento. Las condiciones de la tiroides en el síndrome de DICER1, que se describen a continuación, no alteran la actividad de la hormona tiroidea.
En el síndrome de DICER1, la función de control de la hormona de la glándula no se ve afectada, pero pueden aparecer nódulos benignos (tumores) en la glándula, provocando un agrandamiento con inflamación visible en la parte delantera inferior del cuello, denominado “bocio”. Rara vez los nódulos son malignos.
Bocio multinodular
La anormalidad más frecuente en el síndrome de DICER1 es el desarrollo de múltiples nódulos benignos (nódulos y quistes) dispersos a lo largo de la glándula. Cuando son suficientemente grandes como para notarse, forman lo que se llama “bocio multinodular”. Por lo general, ambos lados de la glándula están afectados. Los nódulos no son malignos. La actividad de la hormona no se altera. Estos bocios son notablemente más frecuentes en portadores de la mutación femeninos que masculinos. Se estima que una gran proporción de los portadores de la mutación de sexo femenino y una proporción mucho menor de portadores del sexo masculino desarrollarán bocio multinodular en el transcurso de sus vidas.
Esta condición (bocio multinodular) es muy común en la población en general. Por lo tanto, el bocio multinodular, en sí, no sugiere una mutación de DICER1 o síndrome de DICER1. Sin embargo, los portadores de las mutaciones tienden a desarrollar bocio multinodular a edades más tempranas que las personas de la población en general. El bocio multinodular en los portadores de la mutación de DICER1 tiende a desarrollarse de los 10 a los 30 años, aunque puede ocurrir tan temprano como a los 5 años.
Carcinoma diferenciado de tiroides
El cáncer diferenciado de tiroides (o “carcinoma diferenciado de tiroides”) (DTC) ocurre raramente en el síndrome de DICER1. En esta condición, los nódulos de tiroides se desarrollan como con el MNG descrito anteriormente, pero el tejido es maligno en lugar de ser benigno. La palabra “diferenciado” se utiliza para indicar que el tejido maligno no se ve primitivo; un tejido primitivo puede indicar una neoplasia agresiva. El DTC no es un cáncer agresivo. Por lo general, es de “grado bajo”, que significa que tiende a permanecer confinado a la glándula tiroides y se puede curar con cirugía.
¿Como son el bocio multinodular o el carcinoma diferenciado de tiroides diagnosticados?
El bocio multinodular y el cancer diferenciado de tiroides tienden a notarse como una holgura en la parte inferior del cuello (“bocio”). Con un examen físico cuidadoso, los médicos pueden detectar nódulos en la tiroides (bocio multinodular o cancer diferenciado de tiroides) antes de que se hagan notorios, como un bocio. La ecografía es una prueba muy fiable y no invasiva que permite definir cualquier anomalía de la tiroides y puede revelar diferencias entre bocio multinodular y cancer diferenciado de tiroides. Las biopsias por punción son necesarias para determinar la verdadera naturaleza de los nódulos de la tiroides.
¿Como son el bocio multinodular o el carcinoma diferenciado de tiroides tratados?
El bocio multinodular puede observarse sin un tratamiento específico, si los nódulos son pequeños y no se agrandan. Si la glándula está agrandada por nódulos múltiples de bocio multinodular o carcinoma diferenciado de tiroides, la glándula tiroides se extirpa quirúrgicamente. La hormona tiroidea se reemplaza exitosamente con medicación diaria.
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
El MNG y DTC son característicos de la mutación de DICER1, pero también son bastante frecuentes en la población en general. Por lo tanto, ninguno de estos diagnósticos en sí es un motivo para realizar pruebas de mutación.
Por otro lado, cuando se da MNG o DTC y cualquier otra enfermedad relacionada con el síndrome de DICER1 en una persona o entre sus familiares cercanos, es razonable que se realice una prueba de detección de mutación de DICER1. El MNG y el tumor ovárico de células de Sertoli-Leydig que ocurren juntos son particularmente sugestivos de la mutación de DICER1.
Blastoma pituitario en el síndrome de DICER1

Algunos tumores cerebrales inusuales ocurren con el síndrome de DICER1. Varios de ellos se discuten en otra sección. Aquí se discute un tumor cerebral, el blastoma pituitario, que es particularmente inusual y especialmente característico del síndrome de DICER1.
¿Que es la glándula pituitaria?
La glándula pituitaria es una glándula pequeña en la parte inferior del cerebro, en el centro de la cabeza, y justo detrás de la pared posterior de las fosas nasales. La glándula pituitaria envía a las hormonas de control central al torrente sanguíneo que, a su vez, controlan muchos otros órganos productores de hormonas como la tiroides, los ovarios y testículos, la glándula suprarrenal, entre otros.
¿Que es el blastoma pituitario ?
El blastoma pituitario es un tumor de la glándula pituitaria que parece ser casi exclusivo del síndrome de DICER1. Es extremadamente raro y solo fue descrito en la literatura médica en 2008, y se vinculó a las mutaciones de DICER1 en 2014. Se han sido reconocido y divulgado menos de 20 casos en la literatura médica mundial. La palabra “blastoma” es un término usado por los patólogos que observan en el microscopio una clase particular de tejido que se parece al tejido de un órgano particular de un embrión humano en etapa temprana.
No se sabe si el blastoma pituitario es maligno o benigno, pero debido a su ubicación en el centro de la cabeza, y como afecta a niños pequeños, se trata de un tumor grave.
El blastoma pituitario aparece en niños muy pequeños; el caso más antiguo reconocido se dio en un niño de 2 años. El tumor puede agrandar la glándula pituitaria y puede protruir por encima de la glándula pituitaria.
¿Cuales son los síntomas del blastoma pituitario?
El blastoma pituitario parece causar, más frecuentemente, un disturbio hormonal que conduce a un aumento de hormonas suprarrenales que causan aumento de apetito y aumento de peso en un patrón particular llamado enfermedad de Cushing, la clase de aumento de peso que se da en personas que deben utilizar “esteroides” para el tratamiento médico. Estos niños ganan peso lentamente durante varios meses, lo que sugiere que el tumor ha crecido lentamente. El blastoma pituitario también puede causar incoordinación de los movimientos oculares. Otros síntomas pueden incluir fatiga, crecimiento más lento, alteración de las vías de fluidos en el cerebro (“hidrocefalia”) y otros.
¿Como es el blastoma pituitario tratado?
Los síntomas del blastoma pituitario son generalmente muy pronunciados, lo que lleva a los médicos a una investigación intensa. El blastoma pituitario es tan raro que los médicos rara vez consideran la posibilidad hasta que la investigación está bien avanzada. Una tomografía computada (CT) o una imagen por resonancia magnética (MRI) de la cabeza revela fácilmente un tumor. La cirugía puede eliminar o disminuir notablemente el tamaño del tumor. No se sabe si es necesaria la terapia luego de la cirugía del blastoma pituitario. Algunas personas, aparentemente, han vivido durante muchos años y, aparentemente, se han curado. Los desequilibrios hormonales se tratan con diversas terapias de reemplazo hormonal.
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
Como el blastoma pituitario es muy característico del síndrome de DICER1, se recomienda realizar pruebas de detección de mutaciones de DICER1 para cualquier niño que presente este tumor.
Quistes y tumores renales en el síndrome de DICER1
Algunos tumores cerebrales inusuales ocurren con el síndrome de DICER1. Varios de ellos se discuten en otra sección. Aquí se discute un tumor cerebral, el blastoma pituitario, que es particularmente inusual y especialmente característico del síndrome de DICER1.
¿Como estan afectados los riñónes con el síndrome de DICER1?
Pueden ocurrir varias enfermedades del riñón debido a las mutaciones de DICER1. La más frecuente es el “nefroma quístico” que afecta alrededor del 5 al 7 % de los niños con la mutación de DICER1. Además, pueden darse dos tumores malignos diferentes.
Nefroma Quístico
El nefroma quístico es una anomalía benigna que puede ocurrir en uno o en ambos riñones de los niños con una mutación de DICER1. Es una de las enfermedades más frecuentes en el síndrome de DICER1. El nefroma quístico es una enfermedad en la que se presentan quistes redondos, tipo globo y llenos de líquido en medio del tejido renal normal. Estos quistes pueden ser pequeños, alrededor de ½ a 1 pulgada de diámetro, o puede haber grupos de quistes muy grandes que pueden alcanzar varias pulgadas de diámetro. Los quistes pueden deformar y, a veces, reemplazar el tejido normal del riñón. A veces, el nefroma quístico se denomina “tumor” porque, médicamente, “tumor” significa nódulo o masa; sin embargo, el nefroma quístico no es canceroso ni maligno.
¿Cuando ocurre el nefroma quístico y como es tratado?
El nefroma quístico casi siempre aparece en niños, entre el nacimiento y los 4 años; es muy raro que se produzca más adelante, tal vez hasta la edad de 12 a 15 años. Como los niños pequeños no nos pueden decir exactamente lo que están sintiendo, no se conoce si el nefroma quístico causa dolor, pero probablemente el dolor no sea un problema importante con el nefroma quístico. Por lo general, el nefroma quístico llama la atención de los médicos porque los padres notan una “masa” o abultamiento en el abdomen. Algunos quistes del nefroma quístico son tan pequeños que se descubren casualmente cuando a un niño se le realizan rayos X u otros diagnósticos por imagen, por otros motivos.
Por lo general, el nefroma quístico afecta un riñón, pero no es especialmente extraño que afecte ambos riñones. La cirugía se utiliza para curar el nefroma quístico. Un nefroma quístico pequeño se puede quitar, al sacar una pequeña porción del riñón. Es poco común que los quistes se desarrollen después de quitar el nefroma quístico. Si un nefroma quístico grande deforma demasiado un riñón, es posible que se deba quitar el riñón entero. A pesar de la extirpación de un riñón, o incluso de un riñón y parte del otro riñón, es extremadamente raro que se comprometa la función renal de un niño después del tratamiento quirúrgico de un nefroma quístico.
Si a un niño con una mutación de DICER1 se le descubren pequeños quistes de riñón por casualidad, es muy probable que sean nefroma quístico. Solo se puede hacer un diagnóstico preciso si se quitan los quistes y se examinan con un microscopio. Sin embargo, cuando los quistes son pequeños y asintomáticos, no son necesarios la extirpación quirúrgica ni un diagnóstico preciso. Aún se desconoce cómo ocuparse mejor de los pequeños quistes. Sin lugar a dudas, algunos médicos recomiendan la extirpación; otros pueden sugerir que se realicen diagnósticos por imagen periódicos.
¿Ocurren tumores renales malignos con mutaciones DICER1?
Es posible que aparezcan dos tumores malignos renales en los portadores de la mutación de DICER1. Afortunadamente, son muy poco comunes.
Tumor de Wilms
El “tumor de Wilms” es un tumor maligno de riñón que se produce en la niñez: la mayoría de los casos ocurre antes de los 10 años, y de los 2 a los 4 años es la edad pico para que se produzca el tumor de Wilms. La mutación de DICER1 no es un factor para más del 95 % de los casos de los tumores de Wilms. Sin embargo, muy pocos niños con una mutación de DICER1 han desarrollado el tumor de Wilms. Si bien es maligno, por lo general, el tumor de Wilms es muy curable.
Sarcoma anaplásico del riñón
Anaplastic sarcoma of the kidney (ASK) is an extremely rare kidney tumor, but it appears to be particularly related to a DICER1 mutation and to cystic nephroma (CN, discussed above). Only since about 2014 has ASK has been recognized to be associated with DICER1 syndrome, so much remains to be learned. ASK may be preceded by cystic nephroma, but the interrelationships between CN and ASK are yet to be learned. CN is one of the more frequent conditions associated with a DICER1 mutation, whereas ASK is one of the rarest. Therefore, it appears that only rarely is CN followed by ASK. ASK can occur under the age of two years, but also up to age 20 years and perhaps beyond. It is malignant and appears to require aggressive treatment, and children have certainly been cured.
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
El nefroma quístico y el sarcoma anaplásico del riñón son característicos de las mutaciones de DICER1. Por lo tanto, si un niño es diagnosticado con nefroma quístico o sarcoma anaplásico del riñón, es recomendable hacer una prueba para detectar mutaciones de DICER1. El tumor de Wilms es tan raramente asociado con mutaciones de DICER1 que el diagnóstico no sugiere una mutación de DICER1. Solo se recomienda realizar una prueba para detectar mutaciones cuando un paciente con tumor de Wilms u otros miembros de la familia tienen una o más de las otras enfermedades asociadas con el síndrome de DICER1 (ver las otras enfermedades discutidas aquí).
Tumor ovárico de células de Sertoli-Leydig y otros tumores del tracto reproductor femenino en el síndrome de DICER1.

Un tumor del ovario (el tumor de células de Sertoli-Leydig) se encuentra entre las manifestaciones frecuentes de la mutación de DICER1 en las mujeres. Otro tumor ovárico y un tumor del cérvix uterino (llamado rabdomiosarcoma embrionario) son manifestaciones muy raras de la mutación de DICER1.
¿Que es el tumor ovárico de células de Sertoli-Leydig?
El tumor ovárico de células de Sertoli-Leydig (TCST) es uno de los tumores más frecuentes que ocurren en el síndrome de DICER1. Es una forma rara de tumor ovárico, que comprende a menos del 5 % de todos los tumores ováricos de la población en general, pero el TCST es bastante característico del síndrome de DICER1. No se conoce el porcentaje de mujeres portadoras de la mutación que desarrollan el TCST, pero una estimación es que probablemente menos del 10 % de las mujeres portadoras desarrollen TCST. El TCST es maligno, pero generalmente no es una neoplasia agresiva, ya que la mayoría de los pacientes se curan con la extirpación quirúrgica del tumor.
El TCST ovárico no está relacionado de modo alguno con los tumores ováricos malignos más comunes que ocurren en la población en general. Estos tumores ováricos pueden ocurrir en mujeres con una predisposición al cáncer de mama. Una vez más, no existe una conexión entre la predisposición de tumor ovárico y cáncer de mama, y el síndrome de DICER1, y, conforme al conocimiento actual, los portadores de la mutación de DICER1 no tienen una susceptibilidad mayor al cáncer de mama.
El TCST es uno de los miembros de una familia de tumores ováricos denominados “tumores de células estromales”. Otros tumores de células estromales ocurren muy raramente en el síndrome de DICER1. El “ginandroblastoma” y el “tumor de células de la granulosa tipo juvenil” son ejemplos de tumores de células estromales raros que se han producido en el síndrome de DICER1.
¿Cuando ocurre el TCST y cuales son sus síntomas?
En el síndrome de DICER1, el TCST tiende a ocurrir entre los 10 y los 30 años, aunque raramente puede ocurrir en niñas de dos a cinco años de edad, o en mujeres menores de 50 años.
Los síntomas de TCST pueden deberse a las hormonas que puede producir el TCST, con un aumento del vello facial o corporal, agravamiento de la voz o cambios menstruales. Es posible que no haya ningún cambio hormonal y el tumor se nota por un abultamiento abdominal (“una masa en el abdomen”) o por cambios en los patrones del intestino y la vejiga.
El TCST ovárico en el síndrome de DICER1 puede ocurrir raramente en ambos ovarios. Cuando sucede esto, cada tumor es un caso diferente y tienden a ocurrir con pocos meses o años de diferencia, uno del otro.
¿Que es rabdomiosarcoma embrionario ovárico?
El rabdomiosarcoma embrionario ovárico (REO) es un tumor muy raro y maligno que ocurre en el síndrome de DICER1. Tiende a ocurrir entre los 10 y los 30 años. Solo se conoce que hayan ocurrido unos pocos casos. El nombre “rabdomiosarcoma embrionario” se refiere a un cierto aspecto de un tumor que se observa con microscopio. Este aspecto del REO es un hilo común en varios tumores de DICER1, en varias partes del cuerpo.
El REO ovárico llama la atención de los médicos debido al abultamiento en el abdomen o quizás debido a los cambios menstruales. Se realiza la extirpación quirúrgica del ovario. Como se han divulgado muy pocos casos, no existe ningún patrón establecido de cuidado, por lo tanto, los oncólogos decidirán si se necesita más terapia en base a las circunstancias únicas del paciente.
¿Que es rabdomiosarcoma embrionario ovárico?
El REO también puede desarrollarse como un tumor del cérvix uterino y es una manifestación muy rara de la mutación de DICER1. Tiende a aparecer entre los 10 y los 20 años y pocas veces después, y se presenta como masas de tumor en la vagina y causa un sangrado leve que se confunde fácilmente con el sangrado menstrual anormal. Cuando aparecen las masas de tumor en la vagina, el REO cervical se conoce por un nombre especial: “Sarcoma botrioide del cérvix”.
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
El TCST y algunos otros tumores ováricos de células estromales en niñas y mujeres jóvenes son muy característicos de la mutación de DICER1, pero también ocurren en la población en general. Si una mujer es diagnosticada con uno de estos tumores, se recomienda realizar una prueba para detectar mutaciones de DICER1.
El REO ovárico y del cuello uterino son característicos del síndrome de DICER1 y es muy raro que aparezcan en la población en general. Si una mujer es diagnosticada con uno de estos tumores, se recomienda realizar una prueba para detectar mutaciones de DICER1.
Varios tumores poco comunes en el síndrome de D1

Una de las características generales más singulares del síndrome de DICER1 es que los portadores de la mutación son susceptibles a condiciones muy inusuales. Si bien estas enfermedades son raras en general, no obstante ocurren en este síndrome con lo que parece estar aumentando su frecuencia. Aunque pareciera que ocurren con más frecuencia en el síndrome de DICER1 que en la población en general, estas condiciones siguen siendo poco comunes en el síndrome.
Además de los tumores poco comunes discutidos en otras secciones, aquí se discuten otras tres enfermedades raras:
- Tumor ocular: “Meduloepitelioma de cuerpo ciliar”
- Tumor del seno nasal: “hamartoma condromesenquimal nasal”
- Pólipos en el tracto intestinal: “pólipos hamartomatosos juveniles”
Meduloepitelioma de cuerpo ciliar
 Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
CBMEs in DICER1 syndrome have occurred in children under 10 years of age. Fewer than 20 cases connected to DICER1 have been reported in the medical literature, so it is very unusual even in DICER1 syndrome
¿Como se diagnostica el meduloepiteloma de cuerpo ciliar?
Como este tumor se presenta en la parte delantera del ojo, a veces los padres lo notan como un aspecto inusual de la pupila del ojo. Además, se pueden detectar problemas en una prueba de visión escolar. La visión en un ojo puede deteriorarse significativa o parcialmente.
Un oftalmólogo notará fácilmente una anormalidad a través de un examen ocular regular y las exploraciones por CT o MRI detectarán cambios en la parte delantera del globo ocular. Por lo general, se consultará a un médico muy especializado, quien reconocerá la anomalía del ojo como un posible MECC.
¿Como se trata el meduloepiteloma de cuerpo ciliar?
Todo tumor ocular en un niño requiere la atención de oftalmólogos altamente especializados. Según el tamaño del tumor y el grado de deterioro de la visión, el ojo deberá quitarse. Sin embargo, a veces, el ojo puede salvarse a pesar de tener una visión reducida
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
El MECC es un tumor muy poco común y, al parecer, ocurre con excesiva frecuencia en las personas con mutaciones de DICER1. El MECC también ocurre en personas sin mutaciones de DICER1. Por lo tanto, si un niño es diagnosticado con MECC, se recomienda investigar cuidadosamente a la familia del niño para descubrir si se conocen otras enfermedades asociadas con la mutación de DICER1. Si existieran tales enfermedades, entonces se recomienda realizar pruebas de detección de DICER1 al niño con MECC (o a las personas con otras enfermedades). Si no se registran en la familia otras enfermedades asociadas con el DICER1, no es tan urgente la realización de pruebas de detección de mutaciones de DICER1, pero se recomienda hacerlo si la familia lo desea.
Hamartoma condromesenquimal nasal

Como muchos tumores asociados con las mutaciones de DICER1, el hamartoma condromesenquimal nasal (HCEN) es poco común; sin embargo, ocurre a veces en el síndrome de DICER1. Se presenta en la cavidad nasal y en los senos adyacentes a la cavidad nasal. (Los senos son esencialmente callejones sin salida llenos de aire, o “afloramientos”, unidos a la cavidad nasal. Los senos permiten que varios huesos faciales sean más ligeros de peso, al crear bolsillos llenos de aire dentro de ellos).
El HCEN es un tumor benigno, un crecimiento benigno. Es nombrado por los patólogos por su aspecto a la vista del microscopio: “condromesenquimal” significa que el tejido tiene características de cartílago y otros tejidos conectivos. La palabra hamartoma es difícil de definir, pero, por lo general, significa que el material del tumor parece bastante normal, aunque hay demasiado de él.
El HCEN puede ser bilateral, es decir, afectar los senos derecho e izquierdo al mismo tiempo. A medida que se agranda un tumor HCEN, puede empujar hacia los lados y deformar los huesos faciales pequeños, pero los empuja (en lugar de invadirlos), lo que es consistente con el concepto de que el HCEN no es maligno.
¿Como se diagnostica el Hamartoma condromesenquimal nasal?
En el síndrome de DICER1, el HCEN se ha producido en jóvenes de 7 a 20 años, aproximadamente. Se han divulgado menos de 20 casos relacionados con DICER1 en la literatura médica, por lo que es muy inusual incluso en el síndrome de DICER1. En la población en general, el NCMH también es poco común y el diagnóstico puede realizarse a cualquier edad, con una preponderancia de casos en niños menores de 2 años. Las personas con HCEN presentan congestión nasal y “falta de ventilación” como signo de bloqueo o bloqueo parcial de las vías nasales. Los médicos pueden observar tejido inusual en la cavidad nasal. Una exploración por CT o MRI revelan la presencia de tejido blando en la cavidad nasal o en los senos adyacentes. Es necesaria una biopsia quirúrgica para realizar el diagnóstico.
¿Como se trata el Hamartoma condromesenquimal nasal?
Por lo general, la extirpación quirúrgica es exitosa para el HCEN. Pocas veces, puede necesitarse más de una cirugía para quitar todos los HCEN o los HCEN recurrentes (lo cual es poco común).
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
El HCEN, al igual que el CBME que se discutió anteriormente, es poco común tanto en la población en general, como en el síndrome de DICER1. Conforme a la información actual, parece que las personas con mutación de DICER1 tienen una probabilidad ligeramente mayor de desarrollar HCEN. El HCEN también ocurre sin tener una relación con la mutación de DICER1. Por lo tanto, si un niño es diagnosticado con NCMH, se recomienda investigar cuidadosamente a la familia del niño para descubrir si se conocen otras enfermedades asociadas con la mutación de DICER1. Si existieran tales enfermedades, entonces se recomienda realizar pruebas de detección de DICER1 al niño con HCEN (o a las personas con otras enfermedades). Si no se registran en la familia otras enfermedades asociadas con el DICER1, no es tan urgente la realización de pruebas de detección de mutaciones de DICER1, pero se recomienda hacerlo si la familia lo desea.
Pólipos intestinales juveniles

Los pólipos intestinales juveniles son nódulos pequeños del tamaño de una uva o grupos de nódulos del tamaño de una uva que crece desde la superficie interior del tubo intestinal. Pueden bloquear parcialmente el intestino y retardar el paso del contenido intestinal. Son benignos. Es probable que sea solo un pólipo o algunos pocos. Ocurren en la población en general, así como en los portadores del síndrome de DICER1. Son poco comunes en el síndrome de DICER1, si bien aún no hay pruebas de que están directamente conectados con la mutación de DICER1, al parecer sí hay una conexión.
Los pólipos intestinales juveniles no están de ninguna manera relacionados con el síndrome familiar de pólipos en el colon (el “intestino grueso”) ni en el recto; esta condición se llama “poliposis adenomatosa familiar” y está asociada con la susceptibilidad excesiva al cáncer de colon adulto.
¿Como se diagnostican los pólipos intestinales juveniles?
En el síndrome de DICER1, estos pólipos tienden a ocurrir en el intestino delgado y eventualmente bloquean/obstruyen el tracto intestinal causando dolor y distensión abdominal. Las diferentes técnicas de rayos X, como la exploración por CT o un enema de bario (o una técnica similar para niños pequeños) identificarán una obstrucción y pueden mostrar los pólipos dentro del tubo intestinal. Cuando un pólipo se produce en otros lugares del tracto intestinal (tal como el esófago o el recto; algo que ocurre raramente), se utilizarán otros métodos de diagnóstico.
¿Como se tratan los pólipos intestinales juveniles?
Para extraer el pólipo o los pólipos y aliviar cualquier obstrucción intestinal se utiliza la cirugía.
¿Cuando se realiza una prueba para determinar una mutación de DICER1?
Los pólipos intestinales juveniles son poco comunes en el síndrome de DICER1 y, probablemente, son algo más frecuentes en la población en general que en el síndrome de DICER1. Por lo tanto, si un niño tuviera pólipos juveniles y si no hubiera otras enfermedades relacionadas con el DICER1 en la familia, los médicos estarían en lo correcto de no considerar el síndrome de DICER1 y no sería necesario realizar pruebas de mutación de DICER1. Por otro lado, si un niño con pólipos tuviera otras enfermedades relacionadas con el DICER1 o si los familiares cercanos tuvieran tales enfermedades, sería recomendable realizar una prueba de detección de mutación de DICER1.




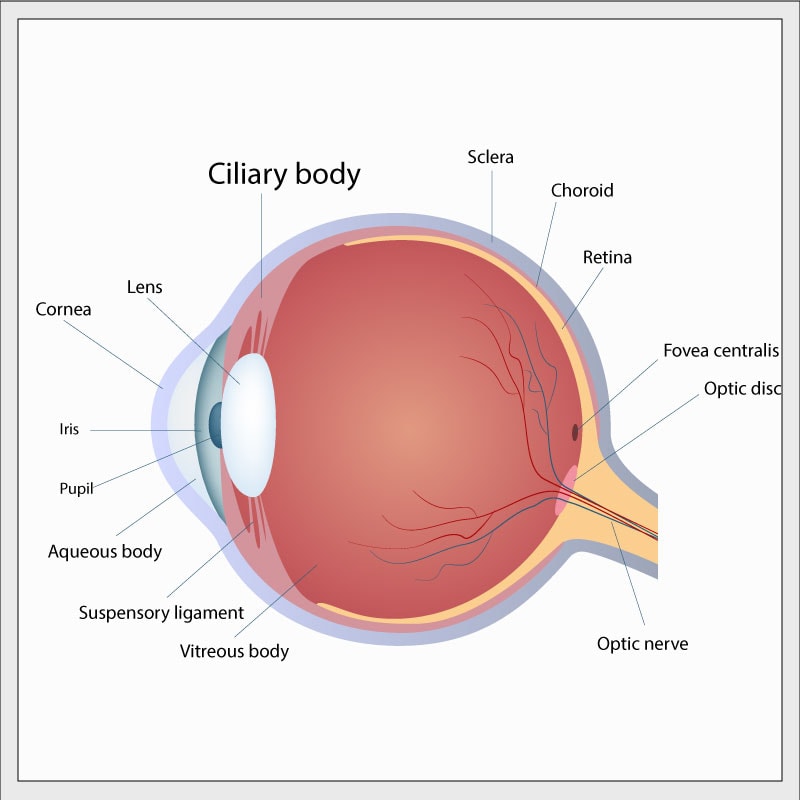 Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.