La présente section est une adaptation de l’article intitulé : « DICER1: mutations, microARNs and mechanisms » publié dans Nature Reviews Cancer par Foulkes, Priest et Duchaine (PMID 25176334). Pour de plus amples informations, voir l’article original.
Pour obtenir une liste récente des articles publiés sur le gène DICER1 et le syndrome DICER1, cliquez ici.
DICER1 : Structure et fonctionSyndrome DICER1 Spectre des tumeurs du syndrome DICER1Prise en charge du syndrome DICER1 : Prévention, dépistage précoce, traitement
DICER1 : Structure et fonction
Fonctions moléculaires
Fonctions moléculaires
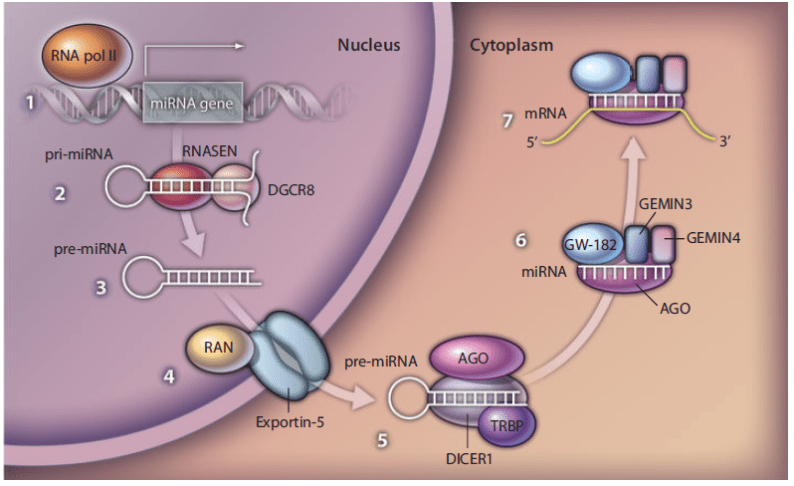
La protéine Dicer est une endoribonucléase à l’origine de la production de micro-ARN (miARN) à maturité qui sont de petites molécules d’ARN monocaténaires qui ciblent les ARN messagers (ARNm) et jouent un rôle important dans la régulation de l’expression des gènes en inhibant la synthèse des protéines. La forme humaine de la protéine Dicer s’appelle DICER1, est codée par le gène DICER1 et est exprimée dans de nombreux tissus. DICER1 joue un rôle dans la voie du silençage de l’expression génique médiée par les miARN, où un miARN s’hybride imparfaitement avec les ARNm cibles. Ceci entraîne généralement la répression de la traduction de l’ARNm et le déclenchement de la décomposition de l’ARNm. Un transcrit primaire de miARN (pri-miARN) est produit par transcription d’un gène de miARN par l’enzyme ARN polymérase II. Le pri-miARN est ensuite transformé dans le noyau par la DROSHA (également connue sous le nom de RNASEN), une endonucléase d’ARN et la protéine de liaison à l’ARN DGCR8 (aussi connue sous le nom de PASHA), ce qui entraîne la production d’une tige-boucle d’ARN qui contient le miARN. Ce mi-ARN prématuré (pré-miARN) en épingle à cheveux est ensuite exporté vers le cytoplasme à travers les pores du noyau. La protéine cytoplasmique DICER1 clive ensuite le pré-miARN pour libérer le miARN court (~ 21 nucléotides) à maturité. Ce miARN est ensuite chargé dans une protéine Argonaute (AGO) qui constitue le cœur du complexe de silençage induit par le miARN (miRISC). Le miRISC s’hybride ensuite avec les ARNm cibles pour produire leur silençage.
Bahubeshi A, Tischkowitz M, Foulkes WD. miRNA processing and human cancer: DICER1 cuts the mustard. Sci Transl Med. 2011 Nov 30;3(111):111ps46. doi: 10.1126/scitranslmed.3002493.
Structure de la protéine DICER1
DICER1 Structure
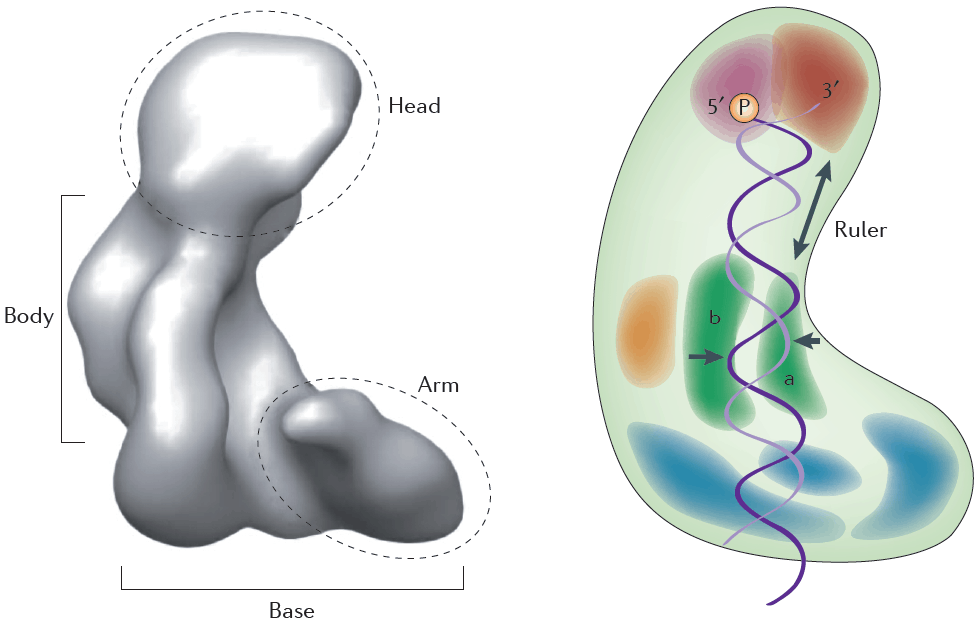
– Le gène DICER1 humain est situé sur le chromosome 14q32.13 et se compose de 27 exons pour un total de 1 922 acides aminés. La protéine DICER1 est une grande enzyme multidomaine en forme de « L ».
Au sommet du « L » se trouvent les domaines PAZ et Platform. Ces domaines sont des poches de liaison pour l’ARN bicaténaire (ARNdb). Le surplomb 3’ du substrat d’ARNdb se lie au domaine PAZ et le surplomb 5’ se lie au domaine Platform.
La moitié inférieure de la protéine DICER1 se compose des domaines RNase IIIa et RNase IIIb, qui se dimérisent pour former le cœur catalytique essentiel de l’enzyme et chacun clive un brin du substrat d’ARNdb. Le domaine RNase IIIa est à l’origine de la production de miARN 3p à partir du brin 3’ et le domaine RNase IIIb produit des miRNA 5p à partir du brin 5’. DICER1 a besoin d’ions de magnésium et/ou de manganèse pour effectuer le clivage catalytique. Ces ions métalliques sont liés à des résidus de liaison des ions dans les domaines RNase III au niveau des résidus d’acide aminé E1320 et E1564 dans le domaine RNase IIIa et D1709 et E1813 dans le domaine RNase IIIb.
Corrélation clinique : La plupart des personnes qui ont des tumeurs liées au syndrome DICER1 se révèlent porteuses de deux mutations de DICER1. Celles-ci consistent généralement en une première mutation hétérozygote tronquante (inactivante) présente dans la lignée germinale qui peut être située à n’importe quelle position dans le gène; la seconde est une mutation somatique faux-sens (spécifique à la tumeur) qui touche toujours l’un des résidus de liaison de l’ion métallique dans RNase IIIb ou un résidu adjacent. Presque toutes les tumeurs et dysplasies associées au syndrome se sont révélées contenir de telles mutations à un point « névralgique ». Dans un très petit sous-groupe de patients atteints du syndrome DICER1, les événements mutationnels initiaux sont des mutations en mosaïque de RNase IIIb. Le phénotype peut être plus grave dans ces cas.
Les domaines RNase III sont séparés des domaines de liaison de l’ARNdb par un lieur qui contient une hélice de raccordement. Il maintient le squelette de phosphate de l’ARNdb en place. Il joue également le rôle d’une « règle » en positionnant le pré-miARN le long de l’enzyme afin qu’elle puisse cliver l’ARNdb en miARN de tailles appropriées.
À la base du L se trouve le domaine hélicase de la boîte DExD/H. On pense que ce domaine maintient en place le substrat d’ARNdb.
Syndrome DICER1
Caractéristiques cliniques/phénotype
Caractéristiques cliniques/phénotype
En 2009, des mutations hétérozygotes DICER1 de la lignée germinale ont été détectées chez onze familles chez lesquelles une ou plusieurs personnes étaient atteintes de blastome pleuropulmonaire (BPP) et avaient des antécédents familiaux de diverses autres tumeurs. Un nouveau syndrome héréditaire de la prédisposition au cancer a ainsi été décrit et appelé par Slade et coll. « syndrome DICER1 » (OMIM n ° 601200).
Depuis lors, le spectre phénotypique a été élargi en répertoriant de nombreuses autres familles comportant des mutations DICER1 de la lignée germinale, y compris des familles avec et sans BPP, et comprend maintenant la prédisposition à d’autres tumeurs, notamment le néphrome kystique (NK), les tumeurs des cordons sexuels et du stroma gonadique ovarien (TCSSGO, en particulier les tumeurs à cellules de Sertoli-Leydig (TCSL)), le goitre multinodulaire (GMN), le rhabdomyosarcome embryonnaire du col de l’utérus (RMSEC) et le blastome pituitaire (BPit), entre autres. Le fait que les deux organes soient touchés n’est pas inhabituel dans le cas des organes pairs. Les mutations de la lignée germinale DICER1 ont été décrites chez des personnes qui présentaient divers autres types de tumeurs et de nouvelles tumeurs continuent d’être ajoutées au spectre du syndrome DICER1. Voir l’onglet « Phénotypes » pour de plus amples informations sur les tumeurs associées au syndrome DICER1.
Beaucoup des tumeurs observées dans le syndrome DICER1 sont assez rares ou inhabituelles. Les tests génétiques de la lignée germinale et la consultation génétique sont suggérés pour un patient et/ou une famille dans l’une des circonstances suivantes : (a) un diagnostic de l’une des tumeurs très caractéristiques (par exemple BPP, NK, BPit); (b) une co-occurrence chez un patient ou une famille de l’une des tumeurs associées au DICER1.
Plusieurs des tumeurs liées à DICER1 sont présentes avant l’âge d’environ 15 à 25 ans et certaines sont présentes très tôt dans l’enfance, comme le BPP, le NK et la BPit. Cependant, la maladie thyroïdienne et certaines autres tumeurs (comme le RMSEC et les TCSSGO) peuvent n’apparaître qu’à 40 ou 50 ans.
La pénétrance des mutations DICER1 pour le développement des tumeurs n’est pas connue avec certitude pour le moment, mais elle semble faible et variable, même au sein d’une même famille. Des études supplémentaires seront nécessaires pour clarifier la pénétrance du syndrome DICER1, car il peut présenter des phénotypes sous-cliniques tels que des kystes pulmonaires ou rénaux ou encore des nodules thyroïdiens minuscules. La pénétrance semble être plus élevée chez les femmes en raison de l’apparition de tumeurs gynécologiques et d’une plus grande fréquence de la maladie thyroïdienne. Il semble qu’au moins 50 % des femmes porteuses et peut-être 80% des hommes porteurs n’ont aucun symptômes cliniques.
Mutations de DICER1 – Cellules germinales et somatiques
DICER1 Mutations – Germline and Somatic
Le syndrome DICER1 est caractérisé par des mutations hétérozygotes de la lignée germinale (c’est-à-dire une mutation sur l’une des deux copies du gène DICER1 se trouvant dans chacune des cellules de l’organisme de chaque personne) et présente une transmission autosomique dominante. Les enfants d’une personne ayant une mutation DICER1 de la lignée germinale ont 50 % de chance d’hériter de la mutation, et donc de risquer d’être atteints des tumeurs liées à la mutation, et 50 % de chances de ne pas hériter de la mutation et ainsi de ne pas présenter de risque accru de tumeurs liées à DICER1. La plupart des mutations DICER1 sont héritées d’un parent qui est porteur de la mutation (qui peut ou non avoir des antécédents personnels de tumeurs liées à DICER1). Dans environ 20 % des cas, les tests familiaux révèlent que la mutation DICER1 est de novo chez la personne concernée. Dans ce cas, il est présumé que les frères et sœurs de la personne concernée ne présentent pas de risque de tumeurs liées à DICER1. Le mosaïcisme gonadique, le phénomène dans lequel un parent n’est pas porteur d’une mutation DICER1 de la lignée germinale, mais possède une mutation DICER1 dans une sous-population de cellules situées dans les gonades, n’a pas été observé dans le syndrome DICER1, mais est théoriquement possible. Par conséquent, des tests génétiques pourraient être envisagés pour les frères et sœurs d’enfants ayant une mutation apparemment de novo, bien qu’un résultat positif soit extrêmement improbable.
Les mutations DICER1 de la lignée germinale sont généralement tronquées (bien que des mutations faux-sens pathogènes aient été observées dans des cas rares) et ont été détectées partout dans le gène. Les mutations DICER1 de la lignée germinale ont été observées dans de nombreux groupes ethniques et aucun effet fondateur n’a été observé. Il n’existe pas de corrélations génotype-phénotype régulières entre les mutations de la lignée germinale et la pénétrance des tumeurs associées.
Le syndrome DICER1 correspond au modèle d’un gène suppresseur de tumeur (GST) dans le sens que le gène est fonctionnellement inactivé par des mutations bialléliques (une mutation de la lignée germinale plus une de la lignée somatique dans le contexte du syndrome DICER1). Cependant, DICER1 diffère de la plupart des autres GST par le fait que la deuxième mutation, ou « deuxième coup », observée dans les tumeurs liées à DICER1 est presque toujours limitée au domaine RNase IIIb. Ces deuxièmes coups somatiques sont très caractéristiques et touchent les positions des acides aminés 1705, 1709, 1809, 1810 et 1813 qui modifient, mais ne détruisent pas, la fonction DICER1. Ces « points névralgiques » sont des résidus qui lient les métaux, qui sont importants pour la fonction enzymatique, ou sont situés adjacents à ces résidus. Les mutations dans ces positions d’acide aminé entraînent la perte de la synthèse de microARN 5p. La perte d’hétérozygotie (PH) est rare chez les tumeurs liées à DICER1 en tant que deuxième événement somatique, mais se produit parfois, surtout dans le cas du pinéoblastome.
Le mosaïcisme somatique pour les mutations des points névralgiques dans le domaine RNase IIIb a été signalé chez des enfants qui présentaient des phénotypes graves remarquables par leurs multiples tumeurs primaires et, dans certains cas, par un surcroissement. Ces mutations semblent être accompagnées de mutations de troncature somatiques dans l’autre allèle ou d’une PH dans les tumeurs.
Spectre des tumeurs du syndrome DICER1
- Blastome pleuropulmonaire (BPP)
Le BPP est une tumeur rare qui survient lors du développement du poumon chez le fœtus et le bébé. Il est considéré comme la manifestation potentiellement mortelle la plus courante du syndrome DICER1. De la naissance jusqu’à l’âge d’environ deux ans, le BPP se présente généralement sous forme d’une masse kystique (BPP de type I). Entre deux et six ans environ, le BPP peut se présenter sous forme de combinaison de kystes pulmonaires et de tumeur solide (BPP de type II), ou de tumeur purement solide (BPP de type III). Les BPP des types II et III sont des tumeurs hautement malignes dont les taux de guérison sont beaucoup plus faibles que dans le cas du BPP de type I. Le BPP de type I est susceptible de se transformer en types II et III, ce qui démontre l’importance d’un dépistage et d’une résection précoces. Les enfants atteints de BPP peuvent présenter un essoufflement et un pneumothorax en raison de la rupture du kyste. Dans les cas de maladie avancée, ils peuvent en outre présenter une perte de poids et de la fièvre. PMID: 8636815, 25209242, 19556464.
Néphrome kystique (NK)
Le NK est généralement considéré comme une dysplasie rénale bénigne, consistant en plusieurs kystes multiloculaires à paroi mince. C’est l’une des manifestations les plus fréquentes du syndrome DICER1 qui survient généralement avant l’âge de quatre ans chez les porteurs de la mutation et se présente sous forme d’une masse indolore à l’abdomen ou au flanc. Bien qu’il ne soit pas néoplasique, une néphréctomie partielle ou totale peut être nécessaire en raison d’un effet de masse ou d’une réduction de la fonction rénale. Dans des cas très rares, on a observé la transformation du NK en sarcome anaplasique du rein, un néoplasme malin. PMID: 21036787, 17137906, 24481001, 28177962
Goitre multinodulaire (GMN) / cancer de la thyroïde différencié (CTD)
Le GMN se caractérise par un élargissement nodulaire de la glande thyroïde. Le GMN est la manifestation la plus fréquente du syndrome DICER1 et se présente sous forme d’une masse palpable au cou, se présentant généralement de l’enfance au début de l’âge adulte. Le GMN et la maladie thyroïdienne sont courants dans la population en général et, par conséquent, la présence du syndrome DICER1 doit être envisagée dans les cas de GMN uniquement en présence d’autres tumeurs liées à DICER1 chez la personne ou la famille. Le CTD est une manifestation rare du syndrome DICER1 qui a été observée chez les enfants qui ont été fortement traités (chimiothérapie, radiothérapie) pour un diagnostic antérieur de BPP ainsi que chez ceux qui n’avaient aucun facteur de risque de cancer de la thyroïde antérieur. PMID: 21205968, 24617712, 27459524.
Tumeur des cordons sexuels et du stroma gonadique ovarien (TCSSGO)
Les TCSSGO sont un groupe de cancers ovariens non épithéliaux inhabituels qui comprend des tumeurs de la granulosa, des gynandroblastomes et des tumeurs à cellules de Sertoli-Leydig (TCSL). Les TCSL sont le type de TCSSGO le plus fréquemment observé dans le syndrome DICER1. Bien que les TCSL soient des tumeurs malignes, la mortalité associée est faible. Les TCSL présentent fréquemment des signes de virilisation (hirsutisme, changements de voix) ainsi que des caractéristiques typiques d’un néoplasme ovarien, y compris une distension et une douleur abdominales. PMID: 21205968, 21501861, 25844550, 27858560.
Blastome pituitaire (BPit)
Le BPit est une tumeur primitive extrêmement rare de la glande pituitaire qui se présente généralement avant l’âge de 24 mois, souvent avec un syndrome de Cushings ou avec de l’opthalmoplégie et/ou de l’hydrocéphalie. Le BPit est une tumeur potentiellement mortelle. C’est une manifestation très rare, mais très caractéristique du syndrome DICER1. PMID: 24839956.
Pinéoblastome (PinB)
Le PinB est une tumeur neuroectodermique primitive rare de la glande pinéale. Ses caractéristiques de présentation comprennent des symptômes de masse pinéale ou pituitaire et des modifications ophtalmologiques. Le PinB est une manifestation rare, mais caractéristique du syndrome DICER1. Il a tendance à se produire d’autant plus tôt qu’une mutation DICER1 de la lignée germinale est présente. PMID: 25022261.
Rhabdomyosarcome embryonnaire du col de l'utérus (RMSEC)
Le RMSEC est une tumeur embryonnaire rare habituellement diagnostiquée dans l’enfance et l’adolescence. Les caractéristiques de sa présentation comprennent des taches ou des saignements anormaux, et une masse polypoïde vaginale. D’autres rhabdomyosarcomes embryonnaires ont été observés dans le syndrome DICER1, par exemple dans la vessie et l’ovaire. PMID: 22180160.
Médulloépithéliome du corps ciliaire (MECC)
Le MECC est une tumeur oculaire embryonnaire très rare qui peut être bénigne ou maligne. Les caractéristiques de sa présentation comprennent une leucocorie ou une diminution de l’acuité visuelle. Le MECC est une manifestation rare, mais assez caractéristique du syndrome DICER1. PMID: 21156700, 27896549.
Hamartome nasal chondromésenchymateux (HNCM)
L’HNCM est une tumeur bénigne très rare des cavités nasales et des sinus, qui se présente souvent avec une congestion nasale dans l’enfance ou l’adolescence. Le HNCM est une manifestation rare, mais assez caractéristique du syndrome DICER1. PMID: 25118636.
Sarcome anaplasique du rein (SAR)
Le SAR est une tumeur rare et agressive du rein. Il a été observé chez très peu de patients porteurs de mutations DICER1 et peut avoir pour origine un néphrome kystique préexistant. PMID: 26928971, 24481001, 27036314.
D'autres tumeurs
– D’autres tumeurs/dysplasies qui peuvent survenir chez les personnes porteuses de mutations DICER1 de la lignée germinale comprennent la tumeur de Wilms, le neuroblastome, le médulloblastome et les polypes intestinaux hamartomateux juvéniles. La grande majorité des cas ne sont pas causés par des mutations DICER1 et la seule observation de ces caractéristiques chez un patient n’est pas une raison suffisante pour soupçonner un syndrome DICER1. Cependant, le risque de ces caractéristiques semble être accru chez les porteurs de mutations DICER1 et des tests génétiques de dépistage des mutations DICER1 peuvent être indiqués si ces caractéristiques sont présentes dans un contexte où il y a d’autres tumeurs liées à DICER1 dans la famille.
Table de phenotypes telechargable
wdt_ID
Phénotype et fréquence relative
Notation
Ce phénotype à lui seul suggère-t-il une mutation dans DICER1?
Âges approximatifs de susceptibilité, tranche (point culminant)
Malin ou Bénin
Mortalité associée
1
Phénotypes les plus fréquents
2
blastome pleuropulmonaire
BPP
oui
3
BPP type I (kystique)
0 - 24 mois (8 mois)
M
oui, s'il y a progression vers le type II ou III
4
BPP type I (kystique)
12 - 60 mois (31 mois)
M
oui, ~40%
5
BPP type III (solide)
18 - 72 mois (44 mois)
M
oui, ~60%
6
BPP type Ir (kystique)
n'importe quel âge
B ou M
aucune observée
7
goitre multinodulaire
GMN
non
5 - 40 ans (10 - 20 ans)
B
non
8
néphrome kystique
NK
oui
0 - 48 mois (non-déterminé)
B
non (voir sarcome anaplasique du rein)
9
tumeurs à cellules de Sertoli-Leydig de l'ovaire
TCSL
oui
2 - 45 ans (10 - 25 ans)
M
oui, < 5% des cas
10
Phénotypes à fréquence modérée
Detection
Detection
L’identification des mutations DICER1 de la lignée germinale chez les patients et les membres de leur famille est importante pour évaluer le risque de néoplasie future et permet de soumettre les familles à des tests prédictifs pour déterminer quelles personnes présentent un risque de tumeurs liées à DICER1.
En raison de la nature unique et rare de beaucoup des tumeurs reconnues comme étant liées à DICER1, la présence de l’un des types de tumeurs distinctifs (BPP, NK, TCSL, RMSEC, BPit, PinB, HNCM et MECC) justifierait le renvoi à un service de génétique du cancer à des fins de consultation et de réalisation de tests génétiques sur la lignée germinale relativement au gène DICER1. De plus, les antécédents familiaux qui contribuent à la présence de caractéristiques moins distinctives liées à DICER1, tels qu’un GMN précoce ou des tumeurs de Wilms peuvent justifier un renvoi aux services de génétique s’ils sont présents dans des familles où il y a des tumeurs liées à DICER1 chez des proches parents. La présence de plusieurs cas de GMN dans une famille peut être une raison suffisante d’envisager le renvoi à un service de génétique.
La détection d’une mutation DICER1 de la lignée germinale permet aux personnes de recevoir un suivi médical personnalisé et d’envisager le dépistage des tumeurs liées à DICER1. Le dépistage des tumeurs permet de les détecter tôt afin qu’elles puissent être enlevées et traitées avec le moins de conséquences possible. Plus précisément, le dépistage du BPP kystique de type I chez les nouveau-nés porteurs de la mutation peut permettre d’enlever chirurgicalement la tumeur et d’empêcher la maladie de progresser vers le BPP de type II ou III dont le pronostic est beaucoup moins favorable. Cependant, de nombreux phénotypes DICER1 sont généralement non mortels et surviennent au cours d’un intervalle d’âge étendu. Ainsi, les examens de dépistage périodiques pluriannuels et la morbidité potentielle de ces programmes doivent être soupesés.
Les membres de la famille non touchés d’une personne porteuse d’une mutation DICER1 de la lignée germinale peuvent être soumis à des tests de dépistage de la mutation DICER1 familiale pour confirmer ou exclure leur statut de personne à risque. Étant donné que la pénétrance du syndrome DICER1 pour les tumeurs semble être assez faible, le dépistage génétique en cascade des parents peut détecter la mutation DICER1 familiale chez des parents non touchés qui risquent d’avoir des tumeurs ou peuvent bénéficier de cette connaissance qui permet une intervention précoce, par exemple dans le cas des tests et du dépistage génétiques des tumeurs chez les bébés.
Surveillance
Surveillance
Il n’existe pas de lignes directrices établies pour la surveillance des tumeurs dans les porteurs de la mutation DICER1.
Les stratégies de dépistage suivantes ont été suggérées par l’atelier sur la prédisposition au cancer de l’enfance AACR (Schultz KAP et al, PTEN, DICER1, FH and their associated tumor susceptibility syndromes: Clinical features, genetics and surveillance recommendations in childhood. Clin Cancer Res 2017 15;23(12):e76-e82). Les recommandations peuvent varier selon les établissements. Il importe de reconnaître que la plupart des données sont fondées uniquement sur l’avis des experts.
Organe
Types de tumeur observés
Symptômes
Surveillance chez les porteurs connus de la mutation
Poumons
BPP, types I, II, III, Ir
Détresse respiratoire, douleur dans la poitrine, fièvre et perte de poids
– Tomodensitogramme initial du thorax entre 3 et 6 mois.
– Si le résultat est normal, tomodensitogramme initial du thorax entre 2,5 et 3 ans.
– La réalisation d’une radiographie du thorax tous les six mois à partir de l’âge de 8 ans, puis annuellement de 8 à 12 ans, devrait être envisagée.
Reins
NK, SAR, TW
Masse abdominale, douleur, sang dans l’urine
– Envisager de réaliser une échographie abdominale tous les six mois jusqu’à l’âge de 8 ans.
– Annuellement par la suite jusqu’à l’âge de 12 ans, selon les résultats.
Cerveau
PinB, BPit
Pinéoblastome : symptômes de masse pinéale ou pituitaire, changements ophtalmologiques. Blastome pituitaire : syndrome de Cushing, ophtalmoplégie ou diabète insipide.
– Une IRM de surveillance du cerveau peut être envisagée, mais est controversée, car le rapport entre le risque et les avantages n’est pas encore connu et ces tumeurs sont toutes les deux des manifestations rares du syndrome DICER1 (<1 %).
– IRM urgente du cerveau en présence de symptômes de pathologie intracrânienne.
Gynécologique
TCSSGO (TCSL, gynandroblastome), RMSEC, RMSEO
TCSSGO : masse/distension abdominale, irrégularités menstruelles, signes de virilisation.
RMSEC : taches vaginales, masse polypoïde qui s’étend du vagin.
– Envisager de réaliser une échographie pelvique annuelle ou semestrielle (en même temps que la surveillance rénale) tous les 6 à 12 mois de la naissance à l’âge adulte.
Thyroïde
Nodules thyroïdiens, GMN, cancer de la thyroïde différencié
Nodules palpables, symptômes de compression locaux (dysphagie, dysphonie, stridor)
– Envisager de réaliser une échographie de la thyroïde avec détermination de la présence d’adénopathies locales à l’âge de 8 ans.
– Si le résultat est normal, répéter l’examen tous les trois ans.
Yeux
MECC
Apparence inhabituelle de la pupille, trouble de la vue
– Connaissance du risque.
Nez
HNCM
Congestion nasale ou « nez bouché »
– Connaissance du risque.
– ENT avec endoscopie nasale si des symptômes d’obstruction nasale sont présents.
GI
Polypes hamartomateux
Obstruction intestinale – ballonnement, douleur
– Connaissance du risque.
– Évaluation si des symptômes d’obstruction intestinale sont présents.
* Ces recommandations peuvent changer à mesure que d’autres données deviendront disponibles concernant le spectre des tumeurs du syndrome DICER1.
Traitement
Il n’existe aucune preuve qui porte à croire que les tumeurs associées aux mutations DICER1 de la lignée germinale présentent des résultats de traitement qui diffèrent de ceux du traitement de ces mêmes tumeurs lorsqu’elles surviennent sporadiquement.
La plupart des tumeurs du spectre du syndrome DICER1 évoluent de façon relativement bénigne. Cependant, la malignité survient dans certains types de tumeurs liées à DICER1, en particulier le BPP. D’autres sont associés à de la morbidité et/ou à de la mortalité en raison de leur emplacement et de leurs effets sur la croissance (comme le BPit) ou des manifestations cliniques telles que l’androgénisation causée par les TCSL. Étant donné que le syndrome DICER1 est rare, les médecins qui observent des maladies liées à DICER1 devraient envisager de communiquer avec des experts du syndrome pour connaître les dernières recommandations en matière de traitement.