DICER1 Síndrome: Genetica, hereditariedade e principais características clínicas

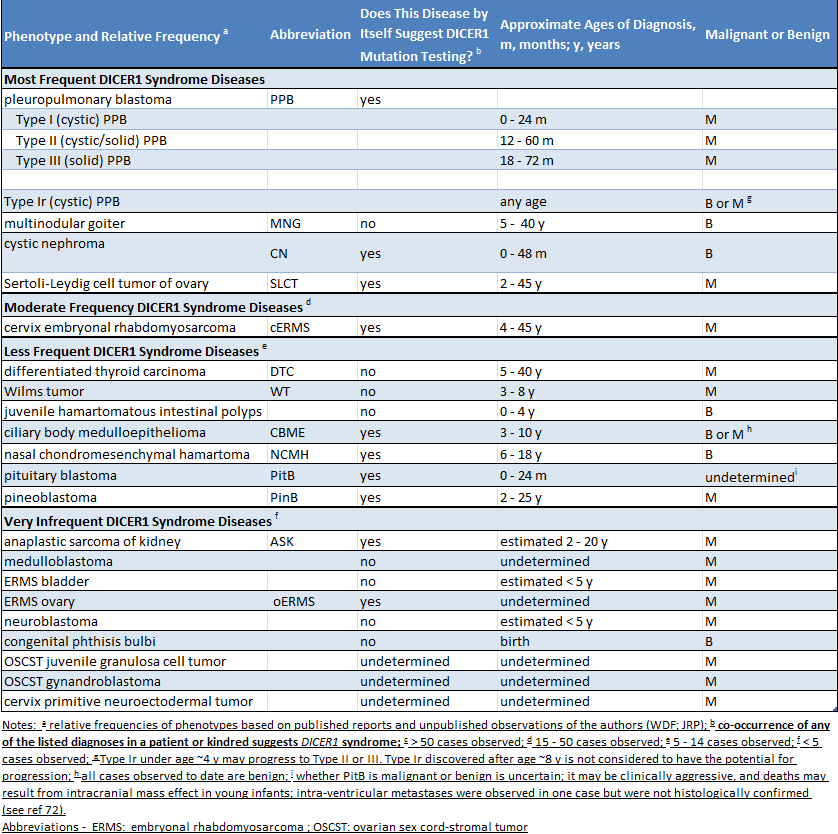
A síndrome DICER1 é uma síndrome familiar de predisposição a tumores que resulta de mutações genéticas que afetam um gene chamado DICER1. Alguns dos tumores relacionados com as mutações de DICER1 são muito graves e ameaçam a vida, mas muitos dos tumores associados a esta síndrome estão longe de ser sérios ou graves. Em geral, os tumores associados a síndrome DICER1 são raros ou muito raros, e alguns são tão raros que podem ocorrer apenas em pessoas com uma mutação DICER1.
Não parece haver nenhum padrão particular no surgimento de tumores, a exceção que cada tipo de tumor associado à síndrome de DICER1 tende a ocorrer em determinadas idades específicas. Em pessoas com esta síndrome, os tumores geralmente ocorrem durante a infância, ou ao redor dos 30 anos de idade. Apesar de indivíduos portadores de mutações no gene DICER1 estarem sob risco de desenvolver os tumores descritos neste site, estima-se que mais da metade das pessoas que carregam mutações em DICER1 nunca irão desenvolver tumores durante a sua vida.
O que é o gene DICER1 e o que ele faz?
Em termos simples, o gene DICER1 indica a cada célula humana quais as instruções genéticas que aquela céula deve obedecer. Especificamente, o gene DICER1 direciona a célula para produzir uma proteína que constitui um mecanismo importante para o controle das atividades celulares. Quase todos os organismos vivos precisam desse tipo de controle e, portanto, a maioria dos organismos vivos tem genes Dicer. “DICER1” é o nome exato da versão humana do gene Dicer, que foi descoberto em um pequeno verme!
Padrões de Herança Genética
Os humanos têm duas cópias do gene DICER1 em todas as células do corpo. Normalmente, a predisposição para a síndrome DICER1 resulta da transmissão de uma cópia anormal (“mutante”) do gene DICER1. Como as células contêm duas cópias de cada gene, as células geralmente funcionam de maneira aceitável mesmo quando uma das cópias é anormal. O gene mutante é transmitido pela mãe ou pai do paciente. O modo de transmissão da síndrome DICER1 é designado pelo nome científico de transmissão “autossômica dominante”. Um dos pais carrega uma cópia normal e uma cópia mutante do gene DICER1. Um filho desse pai herdará a cópia normal ou a cópia anormal; as chances de transmitir uma ou outra dessas versões são de 50%. Cada um dos filhos desse pai tem uma chance em duas de herdá-lo. É provável que metade dos filhos deste pai herde a cópia normal e a outra metade herdará a cópia mutante, e somente os que herdaram a cópia mutnte será suscetível a doenças relacionadas ao DICER1. Essa mutação está presente em todas as células da pessoa e pode ser detectada especialmente no cabelo, na saliva e no sangue. Estima-se que apenas 1 em 10.000 bebês carregam mutações DICER1 no nascimento; seria, portanto, extremamente raro para ambos os pais portarem uma mutação DICER1, e nenhum caso de crianças com mutações DICER1 de ambos os pais foi observado.
Em provavelmente 10 a 15% dos casos, uma mutação DICER1 não é herdada dos pais. É uma nova mutação que ocorre na criança; Não se sabe se a mutação ocorreu em um espermatozóide, no óvulo da mãe ou logo após a concepção. Uma pessoa com uma nova mutação pode transmiti-la para seus filhos.
É possível, mas extremamente raro que a mutação DICER1 esteja presente apenas em certos tecidos do corpo de uma pessoa afetada, por exemplo, em células do pulmão ou células de rim ou de outro tecido específico. As questões de como essas mutações ocorrem ou por que elas afetam apenas certos tecidos permanecem sem resposta. Apenas os tecidos que transportam a mutação são provavelmente afetados por condições relacionadas ao DICER1. Isso é chamado de mutação em “mosaico” porque elas só aparecem em certas células do corpo.
Quais são os sinais de que um indivíduo pode carregar uma mutação DICER1?
Como muitas pessoas com uma mutação em DICER1 não têm problemas de saúde, é muito provável que os pais não saibam que eles carregam essa mutação. A presença de uma mutação em DICER1 pode ser suspeitada em duas situações. Primeiro, se uma criança é diagnosticada com uma doença que está intimamente relacionada com a síndrome DICER1 (como um blastoma pleuropulmonar), uma mutação do DICER1 deve ser procurada. Segundo, se duas ou mais doenças relacionadas ao DICER1 forem diagnosticadas em uma criança ou irmãos ou outros parentes próximos, é razoável buscar uma mutação do DICER1. A tabela a seguir lista as doenças relacionadas a síndrome DICER1. A tabela indica se as condições listadas estão principalmente associadas à síndrome DICER1 (coluna 3, “sim”) ou se ocorrem na população geral e também estão mais raramente associadas à síndrome de DICER1 (coluna 3, “não”)
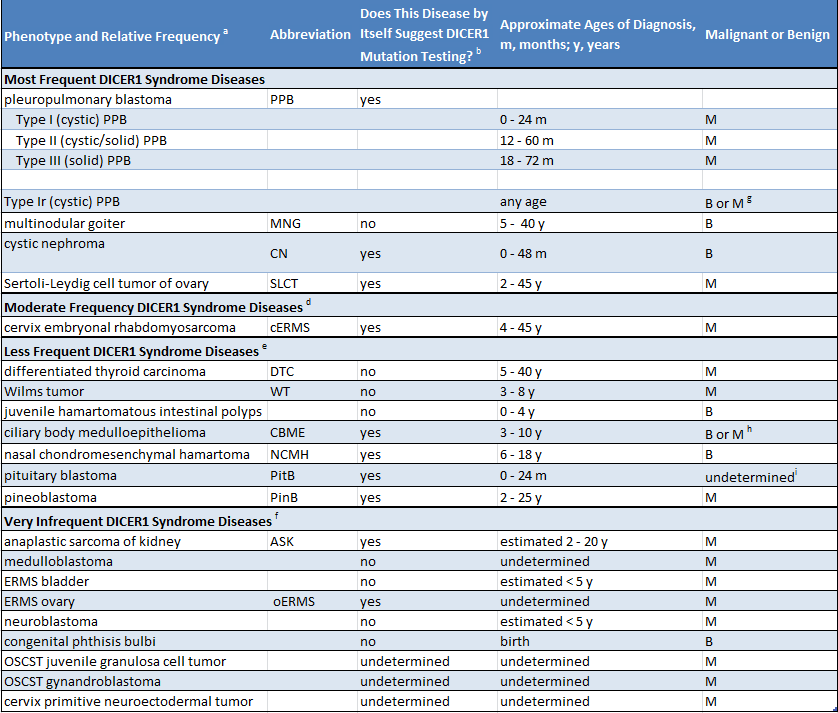
(Clique para ampliar)
Seguem-se exemplos de casos em que se suspeita da presença da síndrome DICER1. Quando uma criança é diagnosticada com blastoma pleuropulmonar (BPP) ou blastoma de hipófise, a suspeita para a síndrome DICER1 é forte porque aproximadamente 75% dos pacientes com BPP são portadores de uma mutação em DICER1, e mais de 90% dos pacientes com blastoma hipofisário têm essa mutação. Por outro lado, por exemplo, se uma criança é diagnosticada com tumor de Wilms (tumor do rim) as suspeitas são baixas porque apenas uma pequena percentagem de pacientes com tumor de Wilms (menos do que 5%) têm uma mutação em DICER1. Certas combinações de doenças (em determinada pessoa ou membros próximos da família) sugerem fortemente a presença da síndrome DICER1. Como exemplos, cerca de 50% dos tumores ovarianos de células Sertoli-Leydig provavelmente são causados por mutações em DICER1, mas quando esse tumor ocorre em associação a nódulos na tireóide (bócio multinodular) no paciente ou em membros da sua família, a combinação do tumor de ovário com o bócio multinodular sugere fortemente que uma mutação DICER1 está causando estas condições.
Mutações do DICER1 em tumores
Por fim, esta discussão sobre a genética do DICER1 deve incluir um fato sobre os genes DICER1 encontrados especificamente em tumores. Os cientistas determinaram que os mesmos tumores geralmente têm duas mutações, uma em cada uma das duas cópias do gene DICER1. A primeira mutação é a “mutação constitucional” da pessoa (está presente em uma cópia do gene em todas as células). A segunda mutação afeta a segunda cópia do gene e está presente apenas nas células tumorais. Nós não sabemos como, porque ou quando a segunda mutação ocorre. No entanto, acredita-se que o tumor se formou porque nenhuma das cópias do gene estava normal nessa condição de duas mutações. Mesmo doenças benignas relacionadas ao DICER1 têm duas cópias mutantes do gene.
Blastoma pleuropulmonar

O blastoma pleuropulmonar (BPP) é uma neoplasia pulmonar rara que ocorre na primeira infância. Foi reconhecido pela primeira vez pelos médicos por volta de 1985. É claro que o BPP existia antes de 1985, mas era tão raro que os médicos nunca o reconheceram como uma doença à parte. A BPP difere completamente e não está relacionada ao câncer de pulmão em adultos. Devido ao fato dessa doença rara ter ocorrido mais de uma vez em algumas famílias, a busca por uma causa levou à descoberta de mutações no DICER1, que agora são conhecidas por causar a grande maioria dos casos de BPP.
Quando o BPP ocorre?
O BPP ocorre quase exclusivamente em crianças menores de quatro anos de idade. Ele também pode ocorrer em bebês. A maioria das crianças com BPP tem uma mutação DICER1, mas casos raros ocorrem na ausência dessa mutação. O BPP apenas muito raramente afeta crianças mais velhas e adultos.
Quais tipos de PPB existem?
O BPP ocorre quase exclusivamente em crianças menores de quatro anos de idade. Ele também pode ocorrer em bebês. A maioria das crianças com BPP tem uma mutação DICER1, mas casos raros ocorrem na ausência dessa mutação. O BPP apenas muito raramente afeta crianças mais velhas e adultos.
O BPP manifesta-se em três formas principais: o primeiro tipo de BPP ocorre em crianças menores de um ano, sendo que o BPP aparece como “cistos” nos pulmões – os cistos da BPP são cavidades cheias de ar que resultam do crescimento anormal dos pulmões. Esses cistos podem ser pequenos e não causar sintomas, enquanto outros podem ser muito grandes e causar dificuldades respiratórias leves ou graves. O BPP cístico é chamado BPP tipo I.
A maioria dos cistos pulmonares que ocorrem em bebês NÃO SÃO BPP; eles são uma condição completamente diferente e menos grave que nada tem a ver com as mutações do DICER1. A cirurgia pode remover cistos pulmonares sem maiores problemas futuros. Como o BPP é raro, e outros cistos pulmonares são mais comuns, os médicos quase sempre consideram os cistos pulmonares como cistos comuns. O diagnóstico de BPP pode ser feito somente após o cisto ter sido removido e depois de examinado ao microscópio. As radiografias do BPP e dos cistos pulmonares mais comuns são semelhantes. A ressecção cirúrgica de cistos do BPP tipo I é geralmente curativa.
A segunda forma de BPP (Tipo II BPP) ocorre geralmente em crianças de 12 a 30 meses de vida. Estes BPPs consistem-se por cistos e massas tumorais sólidas. A terceira forma de BPP consiste em tumores sólidos sem cistos – BPP tipo III – e geralmente ocorre em crianças de 2 a 4 anos. Os tumores tipo II e III da BPP podem causar dificuldades respiratórias, mal estar ou febre e podem apresentar-se como “manchas” na radiografia do tórax; Como o BPP é uma condição rara quando comparado à pneumonia, é comum os médicos pensarem inicialmente que a criança tem pneumonia. Os BPP dos tipos II e III são graves e malignos e requerem cirurgia, quimioterapia e às vezes radioterapia.
Como descrito acima, as faixas etárias dos indivíduos afetados pelas BPP dos tipos I, II e III não são totalmente precisas. Além disso, BPP tipo I pode se transformar em BPP tipo II ou III e, portanto, todos esses tipos estão inter-relacionados e representam um contínuo de manifestações od BPP. Existe uma forma muito rara de BPP do Tipo I, chamada de BPP do tipo Ir, que se acredita ser uma forma não maligna do BPP tipo I e não se transforma em BPP tipo II ou III.
Quando testar para mutação DICER1?
O BPP é muito característico da mutação do DICER1. Portanto, é razoável rastrear as mutações do DICER1 em uma criança diagnosticada com BPP.
Doença Tireoideana na Síndrome DICER1
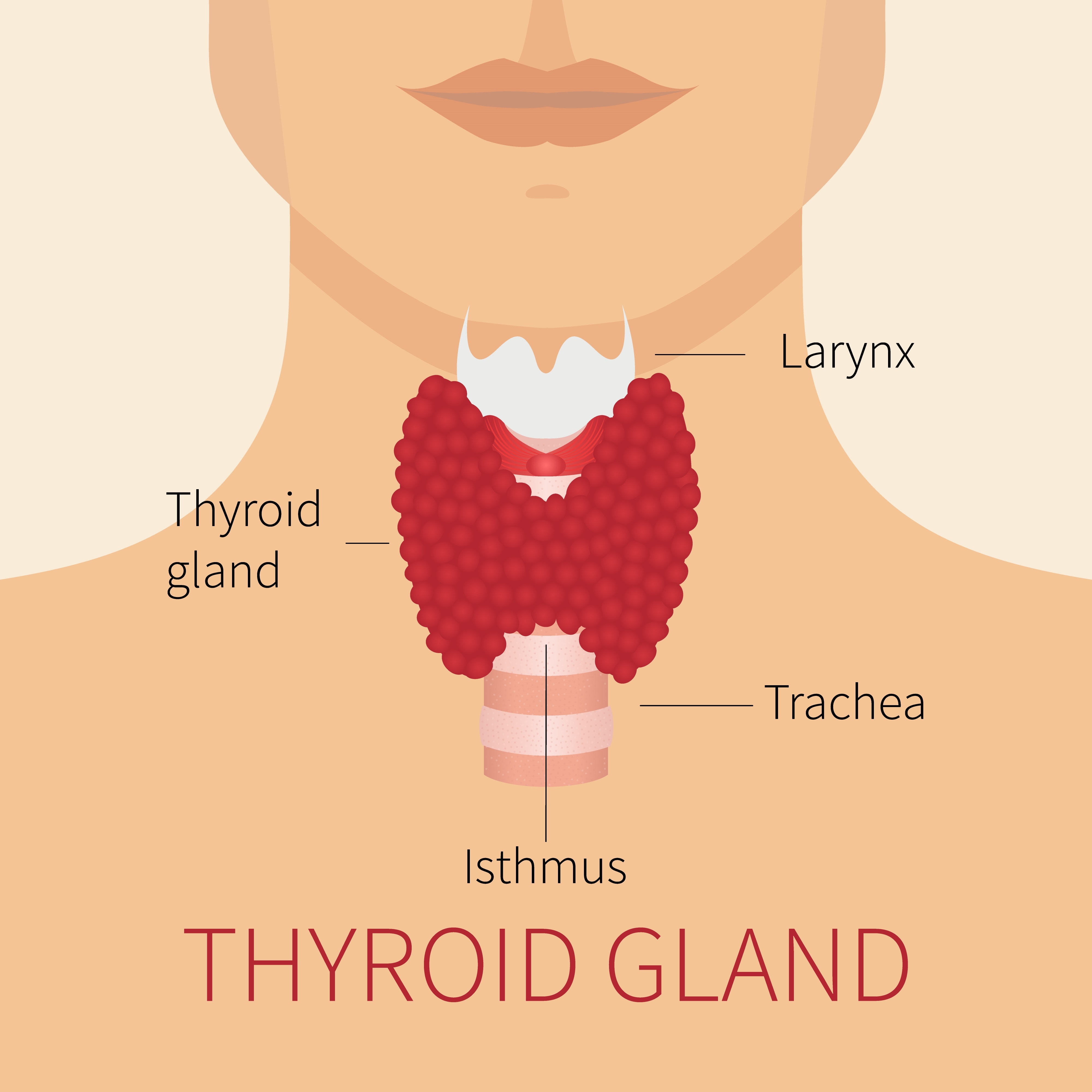
A glândula tireóide é o local das anormalidades mais frequentemente associadas às mutações DICER1. A glândula está localizada na parte anterior do pescoço, logo abaixo da laringe (“pomo de Adão”) e tem lobos em forma de asas de borboleta em cada lado da traqueia, como mostrado na figura ao lado.
A glândula tireóide produz um hormônio que controla muitas ações no corpo, que geralmente são descritas como o ajuste do termostato de energia do corpo. Quando a quantidade de hormônio tireoidiano é muito alta, a pessoa afetada perde peso muito rapidamente e tende a ficar muito quente; quando a quantidade de hormônio tireoidiano é muito baixa, o corpo trabalha em câmera lenta e a pessoa sente frio em excesso, ganha peso o sofre de constipação intestinal. As condições da glândula tireóide encontradas na síndrome DICER1, descritas a seguir, não afetam a atividade do hormônio tireoidiano.
Na síndrome DICER1, a função de controle hormonal da glândula não é afetada, mas massas benignas (tumores) podem se formar na tireóide e fazê-la inchar, surgindo inchaço na parte frontal inferior do pescoço chamado “bócio”. Essas massas de tireóide raramente são malignas.
Bócio Multinodular
A anormalidade mais comum na síndrome DICER1 é o aparecimento de numerosas massas benignas (nódulos e cistos) distribuídas por toda a glândula tireóide. Quando são grandes o suficiente para serem visíveis, formam o que é chamado de “bócio multinodular”. Ambos os lados da glândula são geralmente acometidos. A atividade hormonal da tireóide não é afetada. Esses bócios são muito mais comuns em mulheres portadores de mutações DICER1 do que nos homens. Estima-se que uma grande proporção das mulheres portadoras de mutações em DICER1 e apenas uma proporção muito menor de portadores do sexo masculino desenvolverão bócio multinodular durante sua vida.
Esta condição – bócio multinodular (GMN) – é muito comum na população geral. Portanto, o diagnóstico de GMN não caracteriza a presença de mutações da síndrome DICER1. No entanto, o GMN geralmente ocorre em uma idade mais precoce em portadores de mutação do que em membros da população em geral. Em portadores de mutações DICER1, o GMN geralmente aparece entre as idades de 10 e 30 anos, mas pode ocorrer a partir dos 5 anos de idade.
Carcinoma diferenciado da tireóide
O câncer diferenciado de tireoide (ou “carcinoma diferenciado da tireoide”) (CDT) raramente ocorre relacionado com a síndrome DICER1. No caso desta condição, massas se formam na tireóide do mesmo modo como nos GMN, mas o tecido é maligno em vez de benigno. O termo “diferenciado” é usado para indicar que o tecido maligno não tem aparência primitiva; o tecido primitivo pode indicar a presença de um tumor maligno agressivo. O CDT não é um câncer agressivo. Geralmente é um câncer de “baixo grau”, o que significa que geralmente permanece confinado à glândula tireóide e pode ser curado por cirurgia exclusiva.
Como estas condições são diagnosticadas?
Normalmente, o GMN e o CDT se apresentam na forma de uma protuberância na base do pescoço (“bócio”). Através do exame físico, os médicos podem detectar massas na tireóide (GMN ou CDT) antes de se tornarem perceptíveis na forma de bócio. O ultrasom é um exame extremamente confiável e não invasivo que pode definir a maioria das anormalidades da glândula tireoide e pode revelar diferenças entre o GMN e o CDT. Biópsias de agulha são necessárias para determinar a verdadeira natureza dos nódulos tireoidianos.
Como estas condições são tratadas?
O GMN pode ser apenas observado sem tratamento adicional se os nódulos forem pequenos e não crescerem. Se a glândula tireóide crescer devido à presença de numerosos nódulos de GMN ou CDT, ela geralmente é removida cirurgicamente. A administração diária de medicamentos permite substituir com sucesso o hormônio da tireóide.
Quando testar mutações em DICER1?:
O GMN e o CDT são característicos da mutação DICER1, mas também são relativamente comuns na população geral. Portanto, nenhum desses diagnósticos, por si só, justifica a busca de mutações em DICER1.
Por outro lado, quando um GMN ou CDT e qualquer outra doença relacionada à síndrome DICER1 ocorre em uma pessoa ou em membros próximos da família, é razoável fazer a triagem para as mutações em DICER1. A presença simultânea de um GMN e um tumor de células de Sertoli-Leydig do ovário pode indicar fortemente a existência de uma mutação DICER1.
Blastoma Hipofisário na Sindrome DICER1

Alguns tumores cerebrais raros ocorrem como parte da síndrome DICER1. Discutimos vários deles em outra seção. Um tumor cerebral, o blastoma da glândula pituitária (ou Blastoma hipofisário (BH)), é muito incomum e é particularmente característico da síndrome DICER1. Ele será discutido a seguir.
O que é a glândula pituitária?
A glândula pituitária é uma pequena glândula localizada na parte inferior do cérebro, no centro da cabeça e logo atrás da parede posterior das fossas nasais. A glândula pituitária envia hormônios para a corrente sanguínea, que por sua vez controla vários outros órgãos produtores de hormônios, incluindo a tireóide, ovários, testículos e glândulas supra-renais.
O que é o blastoma da glândula pituitária?
O blastoma da glândula pituitária é um tumor hipofisário que parece estar quase que exclsivamente relacionado à síndrome de DICER1. É extremamente raro e foi descrito na literatura médica apenas em 2008, tendo sido associado a mutações de DICER1 em 2014. Menos de 20 casos foram reconhecidos e relatados na literatura médica mundial. O termo “blastoma” é usado por patologistas que detectam microscopicamente um tipo particular de tecido que se assemelha ao de um órgão em desenvolvimento embrionário inicial.
Não se sabe se o blastoma hipofisário é maligno ou benigno, mas como está no centro do cérebro e afeta crianças pequenas, é considerado um tumor grave.
O blastoma da glândula pituitária ocorre em crianças muito pequenas e o caso de maior idade descrito envolveu uma criança de 2 anos de vida. O tumor pode aumentar a glândula pituitária e pode se projetar acima desta glândula.
Quais são os sintomas do blastoma hipofisário?
Mais comumente, o blastoma da glândula pituitária parece causar um desequilíbrio hormonal que causa um aumento nos hormônios adrenais, resultando em aumento do apetite e ganho de peso de acordo com um padrão específico conhecido por doença de Cushing – o mesmo tipo de ganho de peso observado em pessoas que têm de usar corticoides para fins de tratamento médico. Crianças afetadas ganham peso lentamente ao longo de vários meses, sugerindo que o tumor está crescendo lentamente. O blastoma da glândula pituitária também pode causar incoordenação dos movimentos dos olhos. Outros sintomas possíveis incluem fadiga, desaceleração do crescimento da estatura e interrupção das vias de fluxo de liquor no cérebro (“hidrocefalia”).
Como diagnosticar e tratar o blastoma hipofisário?
Os sintomas do blastoma da glândula pituitária são geralmente muito pronunciados, levando os médicos a investiga-los intensivamente. A extrema raridade do blastoma da glândula pituitária significa que os médicos raramente consideram a possibilidade de sua presença, até que extensa investigação diagnóstica tenha sido realizada. Uma tomografia computadorizada ou ressonância magnética (MRI) da cabeça pode detectar rapidamente um tumor. A cirurgia pode remover o tumor ou reduzir significativamente seu tamanho. Não se sabe se tratamento além da ressecção cirurgica é necessário para o blastoma hipofisário. Alguns pessoas parecem ter apresentado sobrevida longa e foram curadas. Os desequilíbrios hormonais são tratados com diversas terapias de reposição hormonal.
Quando testar mutações em DICER1?
Como o blastoma hipofisário é muito característico da síndrome DICER1, recomenda-se a triagem de mutações DICER1 em qualquer criança com esse tumor.
Cistos e tumores renais na síndrome DICER1
Os rins são os principais órgãos do sistema urinário. Eles exercem várias funções vitais, incluindo a eliminação de toxinas do corpo através da urina. Cistos renais e tumores nesse órgão foram observados em muitos pacientes com síndrome DICER1.
Como a Síndrome de DICER1 afeta os rins?
Muitas doenças renais podem ocorrer devido a mutações DICER1. O mais comum é o “nefroma cístico”, que afeta cerca de 5% a 7% das crianças com mutação DICER1. Além disso, dois tumores malignos distintos podem ser formados.
Nefroma cística
O nefroma cística (NC) é uma anormalidade benigna que pode afetar um rim ou ambos os rins em crianças com uma mutação DICER1. Esta é uma das condições mais comuns na síndrome DICER1. O NC se manifesta nos tecidos normais do rim como cistos arredondados preenchidos com fluido que se assemelha a um balão. Esses cistos podem ser pequenos e de 1 a 2cm de diâmetro, ou podem ser aglomerados muito grandes de cistos que podem ter vários centímetros de diâmetro. Os cistos podem alterar o tecido renal normal e, às vezes, substituí-lo. Às vezes, o NC é referido como um “tumor” porque, do ponto de vista médico, um “tumor” é um nódulo ou massa; no entanto, NC não é tumoral ou maligno.
Quais são os sintomas do nefroma cístico e como é tratado?
O NK quase sempre ocorre em crianças entre o nascimento e os 4 anos de idade; pode ocorrer mais tardiamente, até a idade de 12 a 15 anos, mas isso é raro. Como as crianças pequenas não conseguem explicar com precisão o que sentem, não se sabe se O NC causa dor, mas a dor provavelmente não é um problema significativo para o NC. O NC é geralmente percebido porque os pais percebem a presença de uma “massa” ou nódulo no abdômen. Alguns cistos do NC são tão pequenos que são acidentalmente descobertos quando uma criança é submetida a um raio X ou outro exame de imagem por outras razões.
O NC geralmente afeta apenas um rim, mas não é raro que afete a ambos os rins. A cirurgia é usada como tratamento com objetivode cura para o NC. Um NC pequeno pode ser removido retirando-se uma pequena porção do rim. É incomum que outros cistos se formem após a remoção do NC. Se um NC de grande tamanho deforma um rim, o rim pode ser removido em sua totalidade. Apesar da remoção de um rim ou mesmo de um rim inteiro e de uma porção do outro rim, é extremamente raro que a função renal de uma criança seja comprometida como resultado do tratamento cirúrgico da NC.
Se cistos renais muito pequenos são achados ao acaso em uma criança com uma mutação DICER1, é mais provável que se trate de NC. O diagnóstico preciso só pode ser estabelecido se os cistos forem removidos e examinados ao microscópio. No entanto, pode não haver necessidade de ressecção e diagnóstico preciso quando os cistos são pequenos e não causam nenhum sintoma. O melhor método de tratamento de pequenos cistos é desconhecido atualmente. Enquanto alguns médicos recomendam que sejam removidos, outros podem sugerir a realização periódica de exames de imagem por radiologistas.
Existem neoplasias malignas que afetam os rins no contexto da síndrome DICER1?
Dois tumores renais malignos podem ocorrer em portadores de mutações DICER1. Felizmente, ambos são muito incomuns.
Tumor De Wilms
O “tumor de Wilms” é um tumor renal maligno que ocorre durante a infância – a maioria dos casos ocorre antes dos 10 anos de idade, sendo que esse tumor ocorre mais frequentemente entre os 2 e os 4 anos de idade. A mutação do DICER1 não é um fator associado a esse tumor renal em mais de 95% dos casos de tumor de Wilms. No entanto, alguns poucos casos de crianças portadoras de mutação em DICER1 têm um tumor de Wilms. Embora seja maligno, o tumor de Wilms geralmente é bastante curável.
Sarcoma anaplásico renal
O sarcoma anaplásico renal (SAR) é um tumor renal extremamente raro, mas parece estar particularmente relacionado a mutação no gene DICER1 e ao nefroma cístico (NC, discutido acima). Foi apenas a partir de 2014 que o SAR foi reconhecida como associada à síndrome de DICER1 e, portanto, ainda há muito a ser descrito sobre esta doença. O SAR pode ser precedida pelo aparecimento de nefrose cística, mas as inter-relações entre NC e SAR ainda são desconhecidas. A NC é uma das condições mais comuns emassociação a uma mutação DICER1, enquanto a SAR é uma das condições mais raras. Parece que o NC é apenas raramente seguido por um SAR. O SAR pode ocorrer antes dos 2 anos de idade, mas também até os 20 anos ou mais de vida. Ele é um tumor malignos que frequentemente necessitad de tratamento agressivo, mas com chances de cura.
Quando testar mutações no DICER1?
NC e SAR são doenças bastante características de mutações DICER1. Portanto, se uma criança é diagnosticada com NC ou SAR, é razoável rastrear mutações DICER1. O tumor de Wilms é tão raramente associado com mutações DICER1 que somente o diagnóstico desta condição não sugere a presença de uma mutação DICER1. Apenas quando um paciente tem um tumor de Wilms e outros membros da família tem uma ou mais das doenças relacionadas ao DICER1 (ver outras doenças discutidas aqui), a pesquisa da mutação DICER1 é indicada.
Tumor de células de Sertoli-Leydig do ovário e outros tumores que afetam o sistema reprodutivo feminino na síndrome DICER1

Um tumor ovariano (o tumor de células Sertoli-Leydig) é uma das manifestações frequentes da mutação DICER1 em mulheres. Outro tumor do ovário que também pode acometer o colo do útero (chamado rabdomiossarcoma embrionário) são manifestações muito raras da mutação DICER1.
O que é o tumor de células de Sertoli-Leydig
O tumor de células de Sertoli-Leydig (TCSL) do ovário é uma das manifestações mais comuns da síndrome DICER1. Esta é uma forma rara de tumor ovariano que representa menos de 5% de todos os tumores ovarianos na população geral, mas o TCSL é muito característico da síndrome de DICER1. Nós não sabemos a porcentagem de mulheres com uma mutação em DICER 1 que desenvolvem um TCSL, mas estima-se que provavelmente isso ocorra em menos de 10% delas. O TCSL é maligno, mas geralmente não é um tumor maligno agressivo, e a maioria dos pacientes são curados com a remoção cirúrgica completa do tumor.
Não há ligação entre o TCSL do ovário e as neoplasias ovarianas mais comuns que ocorrem na população em geral. Esses tumores ovarianos podem se formar em mulheres com uma predisposição ao câncer de mama; mais uma vez, não há ligação entre o câncer de mama ou a suscetibilidade a tumor ovariano e a síndrome DICER1, e o entendimento atual sugere que as mulheres com mutações DICER1 não apresentam suscetibilidade aumentada ao câncer de mama.
O TCSL faz parte de uma família de tumores ovarianos denominados “tumores de células estromais”. Outros tumores de células estromais ocorrem muito raramente na síndrome DICER1. “Ginandroblastoma” e “tumor juvenil de granulosa” são exemplos de tumores raros de células estromais que podem surgir na síndrome DICER1.
Quando aparece o tumor de células de Sertoli-Leydig e quais são os sintomas?
Na síndrome DICER1, o TCSL geralmente ocorre entre os 10 e 30 anos de idade, embora raramente ocorra em meninas de dois a cinco anos de idade ou mais tardiamente em mulheres ao redor dos 50 anos.
Os sintomas do TCSL podem ser atribuídos aos hormônios que o TCSL pode produzir e incluem aumento de pêlos corporais ou faciais, engrossamento da voz ou alterações menstruais. Pode não haver alterações hormonais, e o tumor pode ser detectado devido ao inchaço abdominal (“uma massa no abdômen”) ou mudanças nos hábitos intestinais e urinários.
O TCSL do ovário que ocorre na síndrome DICER1 pode ramantente afetar ambos os ovários. Quando isso ocorre, a surgimento em cada um dos ovários de cada tumor é um evento distinto sendo observados com alguns meses ou anos de intervalo entre os tumores.
O que é rabdomiossarcoma embrionário do ovário?
O rabdomiossarcoma embrionário (RMSE) do ovário é um tumor maligno muito raro que ocorre como parte da síndrome DICER1. Geralmente aparece entre as idades de 10 e 30 anos. Apenas alguns poucos casos foram descritos. O nome rabdomiossarcoma embrionário refere-se a uma certa aparência do tumor sob o microscópio. O aparecimento de RMSE é uma característica comum de vários tumores da síndrome DICER1 que afetam diferentes partes do corpo.
O RMSE do ovário é suspeitado pela presença de inchaço abdominal ou por provocar alterações menstruais. O ovário é removido. Como poucos casos foram relatados até o momento, não existe um modelo de cuidado estabelecido, então os oncologistas escolhem o tratamento subsequente com base na situação específica de cada paciente.
O que é rabdomiossarcoma embrionário do colo do útero?
RMSE também pode ocorrer como um tumor do colo do útero e é uma manifestação muito rara da mutação DICER1. Geralmente aparece entre os 10 e 20 anos de vida e raramente após essa idade. Apresenta-se como agrupamentos de tumores na vagina e causa sangramento leve que é facilmente confundido com sangramento menstrual anormal. Quando aglomerados de tumores estão presentes na vagina, o RMSE do colo do útero é por vezes referido como “sarcoma botrióide do colo do útero”
Quando testar mutações no DICER1?
Os RMSE e alguns outros tumores ovarianos de células estromais que ocorrem em meninas e mulheres jovens são muito característicos da mutação DICER1, mas também ocorrem na população geral. Se uma mulher é diagnosticada com um desses tumores, é razoável rastrear a mutação DICER1.
RMSE do ovário e colo do útero são característicos da síndrome de DICER1 e raramente afetam a população em geral. Se uma mulher é diagnosticada com um desses tumores, é razoável rastrear a mutação DICER1.
Miscelânea de tumores raros encotrados na síndrome DICER1

Uma das características gerais mais marcantes da síndrome DICER1 é que os portadores da mutação pode apresentar condições clínicas muito incomuns. Estas condições são geralmente raras, mas, no entanto, ocorrem com frequência aparentemente aumentada nessa síndrome. Embora pareçam ser mais comuns em pessoas com síndrome DICER1 do que na população geral, essas condições também são raramente observadas nessa síndrome.
Além dos tumores incomuns discutidos em outras seções, aqui estão três outras doenças raras:
- Tumor do olho: Meduloepitelioma do corpo ciliar (MECC)
- Tumor do seio paranasal: Hamartoma condromesenquimal nasal (HCMN)
- Pólipos no trato intestinal: pólipos hamartomatosos juvenis
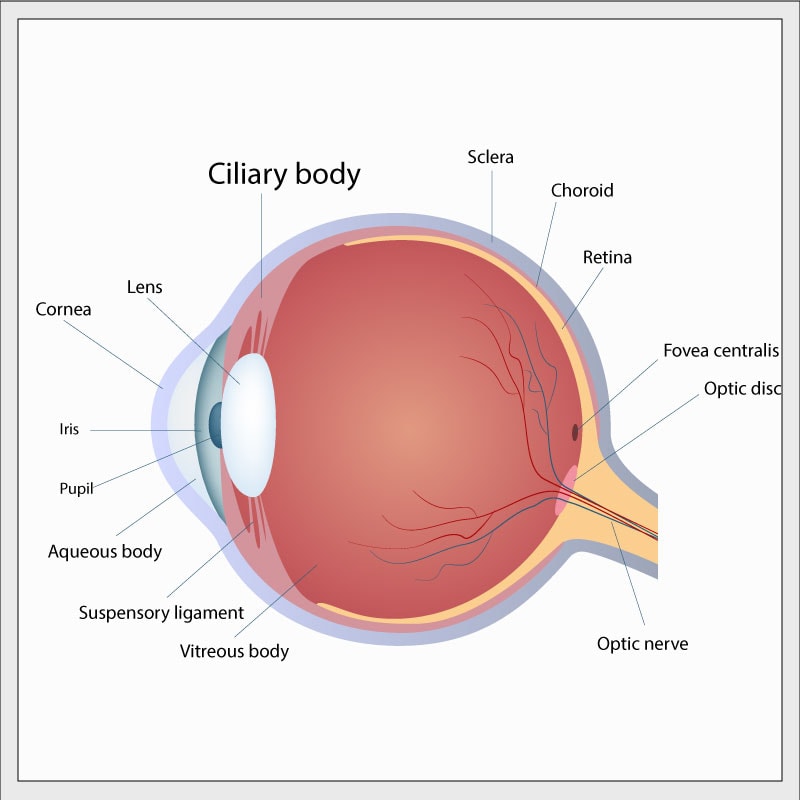
Meduloepitelioma do corpo ciliar

O meduloepithelioma do corpo ciliar (MECC) é um pequeno tumor que afeta a parte frontal do olho. O “corpo ciliar” é uma pequena estrutura complexa em forma de anel e está localizado na parte interna frontal do globo ocular ao redor da córnea. Os músculos do corpo ciliar controlam a forma da pupila, e outras partes do corpo ciliar transportam os vasos sangüíneos usados para alimentar e controlar as partes internas do olho. O termo “meduloepithelioma” é muito complexo e é usado por patologistas para descrever a aparência microscópica de certos tipos de tecido anormal. O MECC pode ser maligno ou benigno – até o momento, os poucos MECCs que foram associados à mutação DICER1 foram benignos.
O casos de MECCs que ocorreram relacionados à síndrome DICER1 afetaram crianças com menos de 10 anos de idade. Menos de 20 casos relacionados ao DICER1 foram relatados na literatura médica e, portanto, MECCs são muito incomuns, mesmo nesta síndrome.
Como o MECC é diagnosticado?
Como esse tumor se forma na parte frontal do olho, sua presença é às vezes notada pelos pais, que percebem o aspecto incomum da pupila do olho. Além disso, problemas podem ser identificados durante um exame oftalmológico na escola. Este tumor pode causar deficiência visual de leve a grave em um olho.
Um oftalmologista notará facilmente uma anormalidade durante o exame oftalmológico periódico, e o uso de uma tomografia computadorizada ou ressonância magnética detectará mudanças parte frontal do globo ocular. Em geral, será consultado um oftalmologista altamente especializado que reconhecerá a possibilidade de que a anormalidade ocular seja um MECC.
Como o MECC é tratado?
A presença de um tumor no olho de uma criança requer a atenção de oftalmologistas altamente especializados. Dependendo do tamanho do tumor e da extensão da deficiência visual, pode ser necessário remover o olho. No entanto, às vezes é possível salvar o olho, apesar poder ocorrer de uma certa diminuição na visão no olho acometido.
Quando testar mutações no DICER1?
O MECC é um tumor muito incomum e parece ocorrer com alta freqência em pessoas com mutações em DICER1. O MECC também ocorre em pessoas que não carregam mutações do DICER1. Portanto, se uma criança é diagnosticada com MECC, é razoável investigar com cuidado para determinar se outras doenças associadas com uma mutação DICER1 foram detectadas na família do paciente. Se assim for, é bastante recomendável rastrear mutações do DICER1 em crianças com MECC (ou em pessoas com outras doenças). Se nenhuma outra doença associada ao DICER1 for identificada na família, o rastreamento de mutações DICER1 é menos necessário, mas é razoável fazê-lo se a família desejar.
Hamartoma condromesenquimal nasal

Como muitos dos tumores associados às mutações DICER1, o hamartoma condromessenquimal nasal (HCMN) é muito incomum, mas às vezes ocorre no contexto da síndrome de DICER1. Este tumor forma-se na cavidade nasal e nos seios adjacentes à cavidade nasal. (Os seios são essencialmente cavidades ósseas cheias de ar, ligados à cavidade nasal. Os seios aliviam o peso de vários ossos do rosto, porque eles criam bolsas cheias de ar dentro de ossos da face).
O HCNM é um tumor benigno – uma massa benigna. Seu nome é atribuído por patologistas por causa da aparência que apresenta durante um exame com microscópio: o termo “Condromesenquima” significa que ele possui tanto características de cartilagem quanto de outros tecidos conjuntivos. Embora o termo “hamartoma” seja muito difícil de definir, geralmente significa que o material do tumor tem uma aparência bastante normal, mas é muito abundante.
O HCNM pode ser bilateral – isto é, ele simultaneamente toca os seios da face direito e esquerdo. À medida que o tumor cresce, ele pode se espalhar e distorcer os pequenos ossos da face, mas não os invade, o que é consistente com o conceito de que o HCNM não é maligno.
Como o HCNM é diagnosticado?
Na síndrome DICER1, o HCNM foi descrito em jovens com idade entre 7 e 20 anos. Menos de 20 casos de HCNM e relacionados ao DICER1 foram relatados na literatura médica; Portanto, é muito incomum, mesmo no contexto dessa síndrome. O HCNM também é muito incomum na população geral; pode ser diagnosticada em qualquer idade e a maioria dos casos ocorre em crianças menores de 2 anos de idade. Pessoas com HCMN têm congestão nasal sinalizando que as passagens nasais estão parcialmente ou completamente obstruídas. Os médicos podem detectar a presença de tecido incomum na cavidade nasal. Uma tomografia computadorizada ou ressonância magnética revela a presença de tecidos moles preenchendo a cavidade nasal ou seios adjacentes, ou ambos. É necessário realizar uma biópsia cirúrgica para estabelecer o diagnóstico.
Como o HCNM é tratado?
A remoção do HCNM é geralmente curativa. Raramente, mais de uma intervenção cirúrgica pode ser necessária para eliminar qualquer HCMN ou HCNM recorrente (o que é incomum).
Quando testar mutações no DICER1?
O HCNM, assim como o MECC que discutimos anteriomente, são muito incomuns na população em geral e no contexto da síndrome de DICER1. Com base nas informações atualmente disponíveis, parece que os indivíduos com uma mutação DICER1 têm um risco ligeiramente maior de desenvolver HCMN. O HCMN também ocorre não relacionado à mutação DICER. Assim, se uma criança é diagnosticada com HCNM, é razoável investigar cuidadosamente para determinar se outras doenças associadas com uma mutação DICER1 foram detectadas na família do paciente. Se assim for, é bastante recomendável rastrear as mutações do DICER1 em crianças com HCMN (ou em pessoas com outras doenças). Se nenhuma outra doença associada ao DICER1 for identificada na família, o rastreamento de mutações DICER1 é menos necessário, mas é razoável fazê-lo se a família desejar.
Juvenile Intestinal Polyps

Os pólipos intestinais juvenis são pequenas massas ou pequenos aglomerados de massas do tamanho de pequenas uvas que se formam na superfície interna dos intestinos. Eles podem causar um bloqueio parcial do intestino e, assim, retardar a passagem do conteúdo intestinal. Eles são benignos. Pode haver apenas um pólipo ou ou vários deles acometendo o intestino. Eles ocorrem na população em geral, bem como na síndrome DICER1. Eles são incomuns na síndrome DICER1 e, embora ainda não haja evidências de que estejam diretamente relacionados a uma mutação DICER1, parece haver um ligação entre a síndrome e estas pólipos intestinais.
Os pólipos intestinais juvenis não estão ligados a síndrome da polipose familiar colônica ou do reto – esta condição é chamado “síndrome da polipose adenomatosa familiar” e está associada com predisposição para o aumento da incidência de cancer do cólon (intestino) no adulto.
Como os pólipos intestinais juvenis são diagnosticados?
Na síndrome DICER1, esses pólipos geralmente se formam no intestino delgado e eventualmente obstruem o trato intestinal, causando dor e inchaço no abdômen. Várias técnicas radiográficas, como a tomografia computadorizada (TC) ou o enema baritado (ou uma técnica semelhante para crianças muito pequenas), podem detectar o bloqueio e revelar a presença de pólipos no interior do trato intestinal. Quando um pólipo é formado em outras partes do trato intestinal (como no esôfago ou no reto, o que raramente acontece), outros métodos diagnósticos são usados.
Como os pólipos intestinais juvenis são tratados?
A cirurgia é usada para remover pólipos e aliviar qualquer obstrução intestinal.
Quando testar mutações no DICER1?
Os pólipos intestinais Juvenis são incomuns na síndrome DICER1 e são, provavelmente, um pouco mais comum na população em geral do que como parte da síndrome DICER1. Portanto, se uma criança teve pólipos juvenis e nenhum membro da família apresenta outras doenças relacionadas com DICER1, os médicos têm razão para não considerar a presença de síndrome DICER1, e seria desnecessário proceder triagem para mutações do DICER1. Por outro lado, se uma criança com pólipos também foi apresenta outras condições relacionadas DICER1 nela mesma ou em membros de sua família, seria razoável para realizar triagem de mutações em DICER1.