DICER1 Syndrome: Genetics, Inheritance and Key Clinical Features
 DICER1 syndrome is a familial tumor predisposition syndrome that results from genetic mutations in a gene called DICER1. Some tumors of DICER1 syndrome are very serious and life threatening, but many of the tumors are not nearly that serious. In general, the tumors are rare or very rare, and some are so rare that they may occur only in individuals with a DICER1 mutation.
DICER1 syndrome is a familial tumor predisposition syndrome that results from genetic mutations in a gene called DICER1. Some tumors of DICER1 syndrome are very serious and life threatening, but many of the tumors are not nearly that serious. In general, the tumors are rare or very rare, and some are so rare that they may occur only in individuals with a DICER1 mutation.
There appears to be no pattern to the development of tumors, except that each tumor in DICER1 syndrome tends to occur at certain typical ages for that tumor. Those who develop the various tumors associated with DICER1 syndrome tend to do so in childhood, even infancy, or by the age of about 30 years old. It is estimated that more than half of the individuals with DICER1 syndrome have no medical problems. While individuals with DICER1 syndrome are at increased risk for developing the tumors recognized in this website, most people with DICER1 syndrome will never develop a tumor.
What is the DICER1 gene and what does it do?
In simple terms, the DICER1 gene is an instruction to every human cell which tells the cell what other genetic instructions to listen to. More specifically, DICER1 gene instructs the cell to make a protein which is a major controller of cell activities. Almost all living organisms require this kind of control so most living organisms have dicer genes. “DICER1” is the specific name for the human version of dicer gene, which was actually first discovered in a small worm!
Inheritance patterns
Humans have two copies of the DICER1 gene in every cell of the body. In general, DICER1 syndrome susceptibility results from inheriting one non-functional (“mutated”) copy of the DICER1 gene. Because cells have two copies of the DICER1 gene, cells generally function acceptably even with one non-functional copy. Individuals typically inherit the mutated DICER1 gene from their mother or father. Scientifically, the inheritance pattern of DICER1 syndrome is called “autosomal dominant” inheritance. A parent has one normal and one mutated copy of the DICER1 gene. A child of this parent will inherit either the normal copy or the mutated copy; the chances are 50-50 which copy of the gene is inherited. Each child of this parent has a 50% chance (1 in 2) of inheriting the working, or non-mutated, copy of the DICER1 gene. These children will not be susceptible to DICER1 syndrome diseases. Each child has a 50% chance (1 in 2) of inheriting the non-working, or mutated, copy of the DICER1 gene, and thus will be susceptible to developing DICER1 syndrome diseases. Such a mutation is present in all the cells of a person’s body and can be detected in hair, saliva, blood, etc. Children can have genetic testing on a blood sample or a saliva sample to see whether they inherited the working or non-working DICER1 gene from their parent. It is estimated that DICER1 mutations are carried by only 1 in 10,000 babies at birth; therefore, having two parents with a DICER1 mutation would be an extremely rare circumstance and no child with DICER1 mutations in both copies of their DICER1 gene, inherited from both parents, has ever been observed.
In probably about 10-15% of circumstances, a DICER1 mutation is not inherited from a parent. Instead, a child has a new mutation; whether the mutation occurred in a sperm cell, in the mother’s egg, or very early after conception is not known. An individual with a new mutation may pass the mutation along to his/her children thereafter.
There are very, very rare individuals in whom a DICER1 mutation is present in only some tissues of their body, for example in lung cells or in kidney cells or in any other specific tissue. How such mutations occur or why they are distributed only to certain tissues are unanswered questions. Only the tissues with the mutation are susceptible to DICER1-associated conditions. These mutations are called “mosaic” because they only appear in some of the cells in the body.
What are the signs that a person may carry a DICER1 mutation?
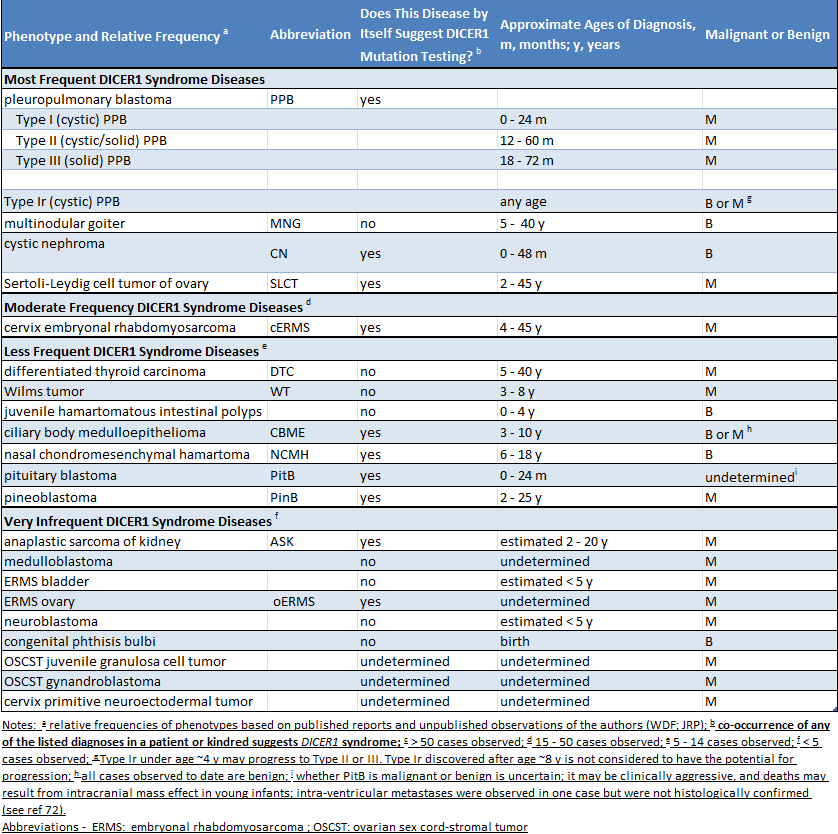
Because many individuals with a DICER1 mutation have no medical problems, it is very possible that a parent is unaware that they carry a mutation. Suspicion of a mutation is raised by either of two circumstances. First, if a child is diagnosed with a disease very strongly connected to DICER1 syndrome (such as pleuropulmonary blastoma), DICER1 mutation should be investigated. Second, if any two or more of the diseases in DICER1 syndrome are diagnosed in one child or in his or her siblings or other close relatives, investigating for a DICER1 mutation is sensible. The following table lists DICER1 syndrome diseases. The table indicates whether the conditions listed are mainly associated with DICER1 syndrome (column 3, “yes”) or whether they occur in the general population and also more rarely in the DICER1 syndrome (column 3, “no”).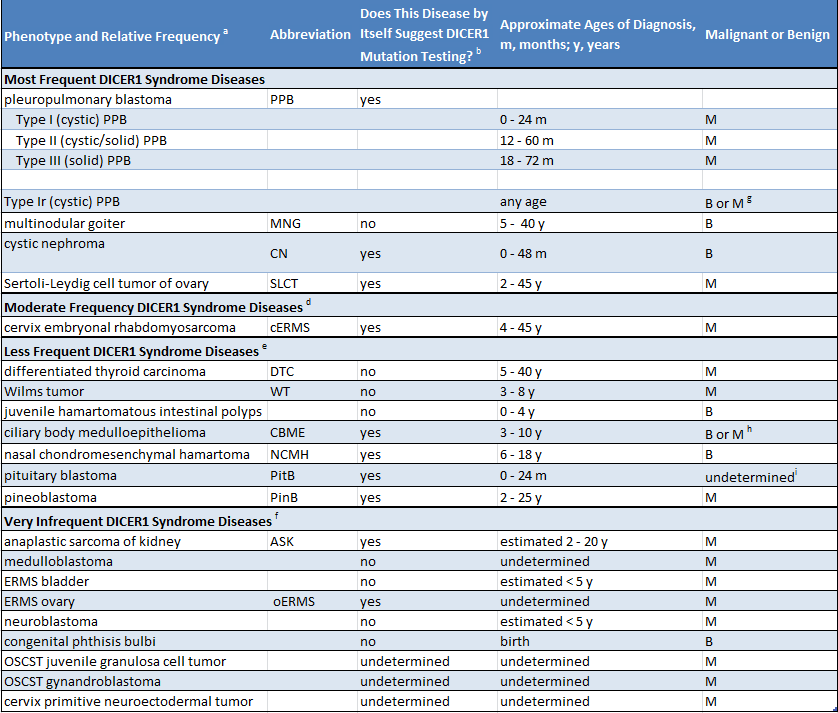
(Click to enlarge)
Examples of when to be suspicious of DICER1 syndrome are as follows. If a child is diagnosed with PPB or pituitary blastoma, the suspicion of presenting a DICER1 mutation is high, because approximately 75% of patients with PPB have a DICER1 mutation and more than 90% of patients with pituitary blastoma have a mutation. In contrast, for example, if a child is diagnosed with Wilms tumor of the kidney, suspicion is not high because only a very small percentage of patients with Wilms tumor (well under 5%) have a DICER1 mutation. Certain combinations of diseases (in one individual or in close family members) strongly raise the suspicion of DICER1 syndrome: for example, probably about 50% of ovarian Sertoli-Leydig cell tumors are caused by DICER1 mutations, but when this tumor occurs with nodules in the thyroid (multinodular goiter) in the patient or in family members, the combination very strongly suggests a DICER1 mutation is causing both conditions.
DICER1 mutations in tumors
Finally, this discussion of DICER1 genetics must include a fact about DICER1 genes in the tumors themselves. Scientists have determined that the tumors themselves usually have mutations in both copies of the DICER1 gene. The first mutation, in one copy of the gene, is the individual’s “constitutional mutation” (present in all cells). The second mutation affects the second copy of the gene and is present only in tumor cells. How, why and when the second mutation occurs is not known. However, it is believed that the tumor developed because neither copy of the gene was normal. Even benign DICER1-associated diseases have two mutated copies of the gene.
Pleuropulmonary Blastoma

Pleuropulmonary blastoma (PPB) is a rare, malignant, early-childhood lung tumor. It was first recognized by doctors in about 1985. Of course, PPB occurred before 1985 but was so rare that doctors had never recognized it as a distinct illness. PPB is completely different from and has no relationship to lung cancer in adults. Because this rare disease occurred in some families, the search for a cause led to the discovery of DICER1 mutations, which are now known to cause the vast majority of PPB cases.
When does it occur?
PPB occurs almost exclusively in children who are under 6 years of age. It may occur in babies. Most children with PPB have a DICER1 mutation, but rarely cases occur without such a mutation. PPB very rarely occurs in older children and adults.
What types of PPB are there?
PPB occurs in three basic forms: first, in children under age 1 year, PPB occurs as “cysts” in the lung – cysts of PPB are air-filled cavities resulting from abnormal lung growth. Such cysts may be small and cause no symptoms, whereas other may be very large and cause mild or severe breathing difficulties. Cystic PPB is called Type I PPB.
Most lung cysts in babies are NOT PPB; they are a completely different and less serious condition and have nothing to do with DICER1 mutations. Surgery removes the cysts without further problems. Because PPB is rare and other cysts are more frequent, doctors almost always consider lung cysts to be the more common type. Only after a cyst is removed and examined under a microscope can PPB be diagnosed. X-rays look the same for PPB and the more common cysts. Surgical removal of PPB cysts is usually curative.
The second form of PPB (Type II PPB) generally occurs in children from age 12 to 30 months. These PPBs are a combination of cysts and lumps of solid tumors. The third form of PPB is solid tumor without cysts – Type III PPB – generally occurring in children from age 2 to 6 years. Types II and III PPB tumors may cause breathing difficulty, general signs of illness or fever with a chest x-ray showing “shadows”; because PPB is rare in comparison to pneumonia, it is usual for doctors initially to think a child has pneumonia. Types II and III PPB are serious and malignant and require surgery and chemotherapy and sometimes radiation therapy. Many children are cured of these tumors but others, unfortunately, may die from these forms of PPB.
As noted above, the age ranges for Types I, II, and III PPB are not precise. Also, Type I PPB can turn into Type II or III PPB so all the Types are connected and represent a “spectrum” of manifestations of PPB. There is a very rare form of Type I PPB called Type Ir PPB which is believed to be a non-malignant form of Type I PPB and does not turn into Type II or III.
When to test for DICER1 mutation?
PPB is very characteristic of DICER1 mutation. Therefore, if a child is diagnosed with PPB, it is reasonable to test for DICER1 mutations.
Thyroid Disease in DICER1 Syndrome
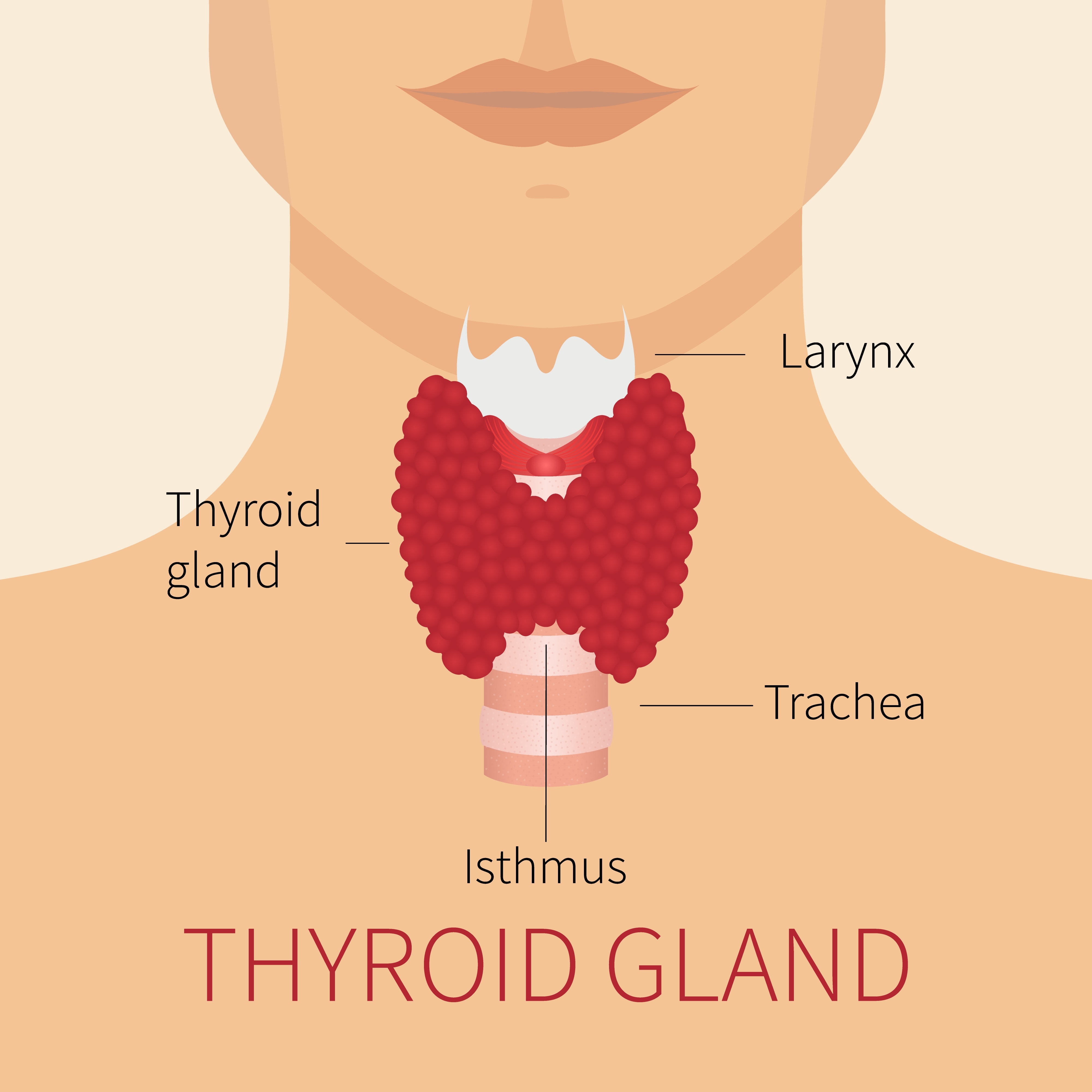 The thyroid gland is the site of the most frequent abnormalities associated with DICER1 mutations. The gland is located in the front of the neck just below the voice-box (“Adam’s apple”) and has butterfly-wing-shaped lobes on each side of the windpipe as shown in the adjacent diagram
The thyroid gland is the site of the most frequent abnormalities associated with DICER1 mutations. The gland is located in the front of the neck just below the voice-box (“Adam’s apple”) and has butterfly-wing-shaped lobes on each side of the windpipe as shown in the adjacent diagram
The thyroid gland produces a hormone which controls many actions in the body which can be generally described as setting the body’s energy thermostat. Too much thyroid hormone and the body goes too fast with weight loss and a tendency to feel too warm; too little thyroid hormone and the body slows down with feeling cold, weight gain and constipation. The thyroid conditions in DICER1 syndrome, described below, do not alter thyroid hormone activity.
In DICER1 syndrome, the hormone control function of the gland is not affected, but benign lumps (tumors) can appear in the gland enlarging it with visible swelling across the lower front of the neck called “goiter”. The lumps are rarely malignant.
Multinodular goiter
The most frequent abnormality in DICER1 syndrome is the development of multiple benign lumps (nodules and cysts) scattered throughout the gland. When they are large enough to be noticeable, they form what is called “multinodular goiter”. Both sides of the gland are usually affected. The nodules are not malignant. The hormone activity is not altered. These goiters are notably more frequent in female rather than male mutation carriers. It is estimated that a large proportion of female and a much lower proportion of male mutation carriers will develop multinodular goiter over the course of their lifetime.
This condition – multinodular goiter (MNG) – is quite common in the general population. Therefore, by itself, MNG does not suggest DICER1 mutation or DICER1 syndrome. However, mutations carriers tend to develop MNG at younger ages than people in the general population develop MNG. MNG in DICER1 mutation carriers tends to develop from age 10 to 30 years although it can occur as early as age 5 years.
Differentiated thyroid cancer
Differentiated thyroid cancer (or “differentiated thyroid carcinoma”) (DTC) rarely occurs in DICER1 syndrome. In this condition, thyroid lumps develop as with MNG described above, but the tissue is malignant instead of benign. The word “differentiated” is used to indicate that the malignant tissue is not primitive looking; primitive tissue can indicate an aggressive malignancy. DTC is not an aggressive cancer. It is usually “low-grade”, which means it tends to remain confined to the thyroid gland and to be curable with surgery.
How are multinodular goiter or differentiated thyroid cancer diagnosed?
Both MNG and DTC tend to be noticed as fullness in the lower neck (“goiter”). With careful physical examination, physicians can detect lumps in the thyroid (MNG or DTC) before they become noticeable as a goiter. Ultrasound examination is an extremely reliable and non-invasive test for defining any thyroid abnormality and may reveal differences between MNG and DTC. Needle biopsies are necessary for determining the real nature of the thyroid nodules.
How are multinodular goiter or differentiated thyroid cancer treated?
MNG may be observed without specific treatment if the nodules are small and not enlarging. If the gland is enlarged by multiple nodules of MNG or DTC, the thyroid gland is surgically removed. The thyroid hormone is very successfully replaced with daily medication.
When to test for DICER1 mutation:
MNG and DTC are characteristic of DICER1 mutation but are also fairly frequent in the general population. Thus, neither of these diagnoses alone is a reason for mutation testing.
On the other hand, when MNG or DTC and any other DICER1-syndrome disease occur in one individual or among their close family members, DICER1 mutation testing is reasonable. MNG and ovarian Sertoli-Leydig cell tumor occurring together are particularly suggestive of DICER1 mutation.
Pituitary Blastoma in DICER1 Syndrome
 A few unusual brain tumors occur in DICER1 syndrome. One brain tumor, pituitary blastoma, is particularly characteristic of DICER1 syndrome, and is discussed here.
A few unusual brain tumors occur in DICER1 syndrome. One brain tumor, pituitary blastoma, is particularly characteristic of DICER1 syndrome, and is discussed here.
What is the pituitary gland?
The pituitary gland is a small gland on the underside of the brain in the center of the head and just behind the back wall of the nasal passages. The pituitary gland sends into the blood stream central control hormones which, in turn, control many other hormone-producing organs such as the thyroid gland, the ovaries and testicles, the adrenal gland and others.
What is pituitary blastoma?
Pituitary blastoma is a tumor of the pituitary gland that appears to be almost unique to DICER1 syndrome. It is extremely rare and was described in the medical literature only in 2008 and linked to DICER1 mutations in 2014. Fewer than 20 cases have been recognized and reported in the world’s medical literature. The word “blastoma” is a term used by pathologists who see a particular kind if tissue under the microscope that looks like the tissue of a particular organ in an early human embryo.
It is not known whether pituitary blastoma is malignant or benign, but because of its location in the center of the head and because it affects small children, it is a serious tumor.
Pituitary blastoma occurs in very young children with the oldest recognized case in a 2-year-old child. The tumor may enlarge the pituitary gland and may bulge up above the pituitary gland.
What are the symptoms of pituitary blastoma?
Most frequently pituitary blastoma appears to cause a hormonal disturbance that leads to increased adrenal hormones which cause appetite gain and weight gain in a particular pattern called Cushing disease – the kind of weight gain seen in individuals who must use “steroids” for medical treatment. These children gain weight slowly over several months which suggests that the tumor has grown slowly. Pituitary blastoma can also cause incoordination of eye movements. Various other symptoms may include fatigue, slower height growth, disturbance of the fluid pathways in the brain (“hydrocephalus”) and others.
How is pituitary blastoma diagnosed and treated?
The symptoms of pituitary blastoma are usually very pronounced which leads physicians to investigate intensively. Pituitary blastoma is so exceptionally rare that physicians will rarely consider the possibility until well into the investigation. A CT or MRI scan of the head readily reveals a tumor. Surgery may be able to remove or markedly diminish the size of the tumor. It is not known whether therapy beyond surgery is necessary for pituitary blastoma. Some individuals appear to have lived for many years and appear to have been cured. Hormonal imbalances are treated with various hormone replacement therapies.
When to test for DICER1 mutation:
Because pituitary blastoma is very characteristic of DICER1 syndrome, testing for DICER1 mutations is strongly indicated for any child with this tumor.
Kidney Cysts and Tumors in DICER1 Syndrome
The kidneys are the main organs of the urinary system. They serve multiple vital purposes including the elimination of toxins that leave the body in the form of urine. Kidney cysts and tumors have been observed in a number of patients with DICER1 syndrome.
How are the kidneys affected in DICER1 syndrome?
Several kidney conditions may occur because of DICER1 mutations. The most frequent is “cystic nephroma” affecting perhaps 5-7% of children with DICER1 mutation. In addition, two different malignant tumors can occur.
Cystic Nephroma
Cystic nephroma (CN) is a benign abnormality which may occur in one or both kidneys of children with a DICER1 mutation. It is one of the more frequent conditions in DICER1 syndrome. CN is a condition of round, balloon-like fluid-filled cysts in the midst of regular kidney tissue. These cysts may be small, such as ½ to 1 inch in diameter, or there may be very large clusters of cysts which may reach several inches in diameter. The cysts may distort and sometimes replace normal kidney tissue. Sometimes CN is called a “tumor” because medically “tumor” means lump or mass – however, CN is not cancerous or malignant.
When does CN occur, what are the symptoms and how is it treated?
CN occurs almost always in children between birth and age 4 years; very rarely it occurs later, perhaps up to age 12 to 15 years. Because young children cannot tell us exactly what they are feeling, it is not known whether CN causes pain, but pain is probably not a major issue with CN. CN usually comes to medical attention because the parents notice a “mass” or bulge in the abdomen. Some CN cysts are so small that they are incidentally discovered when a child has x-ray or other imaging procedures done for other reasons.
CN generally affects one kidney, but it is not especially rare for it to affect both kidneys. Surgery is used to cure CN. Small CN can be removed by taking out a small segment of the kidney. It is unusual for more cysts to develop after CN is removed. If a large CN greatly distorts a kidney, the entire kidney may be removed. Despite the removal of one kidney or even one kidney and part of the other kidney, it is extremely rare for a child to have compromised kidney function after surgical treatment for CN.
If a child with a DICER1 mutation has very small kidney cysts discovered incidentally, they are very likely to be CN. The precise diagnosis can be made only if the cysts are removed and examined under the microscope. However, when cysts are small and asymptomatic, surgical removal and a precise diagnosis may not be necessary. How best to care for small cysts is not currently known. Certainly, some physicians will recommend removal; others may suggest periodic imaging by radiologists.
Do malignant kidney tumors occur in DICER1 syndrome?
Two malignant kidney tumors can occur in DICER1 mutation carriers. Fortunately, each is very unusual.
Wilms tumor
“Wilms tumor” is a malignant tumor kidney tumor of childhood – most cases occur before age 10 years and age 2-4 years is the peak age for Wilms tumor to occur. DICER1 mutation is not a factor for more than 95% of Wilms tumors cases. However, very few children with a DICER1 mutation have developed Wilms tumor. Although malignant, Wilms tumor is generally very curable.
Anaplastic sarcoma of the kidney
Anaplastic sarcoma of the kidney (ASK) is an extremely rare kidney tumor, but it appears to be particularly related to a DICER1 mutation and to cystic nephroma (CN, discussed above). Only since about 2014 has ASK has been recognized to be associated with DICER1 syndrome, so much remains to be learned. ASK may be preceded by cystic nephroma, but the interrelationships between CN and ASK are yet to be learned. CN is one of the more frequent conditions associated with a DICER1 mutation, whereas ASK is one of the rarest. Therefore, it appears that only rarely is CN followed by ASK. ASK can occur under the age of two years, but also up to age 20 years and perhaps beyond. It is malignant and appears to require aggressive treatment, and children have certainly been cured.
When to test for DICER1 mutations:
CN and ASK are characteristic of DICER1 mutations. Therefore, if a child is diagnosed with CN or ASK, it is reasonable to test for DICER1 mutations. Wilms tumor is so rarely associated with DICER1 mutations that the diagnosis does not suggest DICER1 mutation. Only when a patient with Wilms tumor or other family members have one or more of the other diseases associated with DICER1 syndrome (see other diseases discussed here) is mutation testing indicated.
Ovarian Sertoli-Leydig Cell Tumor and Other Female Reproductive Tract Tumors in DICER1 Syndrome
 One tumor of the ovary (Sertoli-Leydig cell tumor) is among the frequent manifestations of DICER1 mutation in females. Another ovarian tumor and a tumor of the uterine cervix (called embryonal rhabdomyosarcoma) are very rare manifestations of DICER1 mutation.
One tumor of the ovary (Sertoli-Leydig cell tumor) is among the frequent manifestations of DICER1 mutation in females. Another ovarian tumor and a tumor of the uterine cervix (called embryonal rhabdomyosarcoma) are very rare manifestations of DICER1 mutation.
What is Sertoli-Leydig cell tumor?
Ovarian Sertoli-Leydig cell tumor (SLCT) is one of the more frequent tumors occurring in the DICER1 syndrome. It is a rare form of ovarian tumor, comprising less than 5% of all ovarian tumors in the general population, but SLCT is quite characteristic of DICER1 syndrome. The percentage of mutation-carrying females who develop SLCT is not known, but an estimate is that probably fewer than 10% of female carriers develop SLCT. SLCT is malignant, but generally not an aggressive malignancy, with most patients cured by surgical removal of the tumor.
Ovarian SLCT is not in any way related to the more common malignant ovarian tumors which occur in the general population. Such ovarian tumors may occur in women with a predisposition to breast cancer; there is no connection between breast cancer-ovarian tumor predisposition and DICER1 syndrome, and, based on current knowledge, DICER1 mutation carriers do not have an increased susceptibility to breast cancer.
SLCT is one member of a family of ovarian tumors called “stromal-cell tumors”. Other stromal-cell tumors occur very rarely in DICER1 syndrome. “Gynandroblastoma” and “juvenile granulosa cell tumor” are examples of the rare stromal-cell tumors that have occurred in DICER1 syndrome.
When does SLCT occur and what are symptoms?
In DICER1 syndrome, SLCT tends to occur between the ages of about 10 to 30 years, although rarely it may occur in girls as young as ages two to five years, or in women up to the age of 50 years.
The symptoms of SLCT may be due to hormones that SLCT can produce, with an increase in body or facial hair, deepening of voice or with menstrual changes. There may be no hormonal changes, and the tumor is noticed by abdominal fullness (“a mass in the abdomen”) or with changes in bowel and bladder patterns.
Ovarian SLCT in DICER1 syndrome may rarely occur in both ovaries. When this happens, each tumor is a different event and such tumors tend to occur a few months or years apart from one another.
What is ovarian embryonal rhabdomyosarcoma?
Ovarian embryonal rhabdomyosarcoma (ERMS) is a very rare, malignant tumor occurring in DICER1 syndrome. It tends to occur between the ages of 10 to 30 years. Only a few cases have been known to occur. The name “embryonal rhabdomyosarcoma” refers to a certain appearance of a tumor under the microscope. This ERMS appearance is a common thread in various DICER1 tumors in various parts of the body.
Ovarian ERMS comes to medical attention because of fullness in the abdomen or perhaps because of menstrual changes. Surgical removal of the ovary is done. Because so few cases have been reported, there is no established pattern of care, so oncologists will decide further therapy based on a patient’s unique circumstances.
What is cervical embryonal rhabdomyosarcoma?
ERMS can also develop as a tumor of the uterine cervix and is a very rare manifestation of DICER1 mutation. It tends to appear between the ages of 10 and 20 years and rarely thereafter and presents as clumps of tumor in the vagina and causes mild bleeding which is easily confused as abnormal menstrual bleeding. When clumps of tumor are present in the vagina, cervical ERMS is sometimes called by a special name “cervix sarcoma botryoides”.
When to test for DICER1 mutation:
SLCT and certain other stromal-cell ovarian tumors in girls and young women are very characteristic of DICER1 mutation but also occur in the general population. If a female is diagnosed with one of these tumors, it is reasonable to test for DICER1 mutation.
Ovarian and cervical ERMS are characteristic of DICER1 syndrome and are very rare in the general population. If a female is diagnosed with one of these tumors, it is reasonable to test for DICER1 mutation.
Miscellaneous Unusual Tumors in DICER1 Syndrome
 One of the most distinctive overall features of DICER1 syndrome is that mutation carriers are susceptible to very unusual conditions. Although these conditions are rare in general, they nevertheless occur in this syndrome with what appears to be increased frequency. Though they seem to occur more frequently in DICER1 syndrome than in the general population, these conditions remain unusual in the syndrome.
One of the most distinctive overall features of DICER1 syndrome is that mutation carriers are susceptible to very unusual conditions. Although these conditions are rare in general, they nevertheless occur in this syndrome with what appears to be increased frequency. Though they seem to occur more frequently in DICER1 syndrome than in the general population, these conditions remain unusual in the syndrome.
In addition to the unusual tumors discussed in other sections, three other rare conditions are discussed here:
- Eye tumor: “Ciliary body medulloepithelioma”
- Nasal sinus tumor: “Nasal chondromesenchymal hamartoma”
- Polyps in the intestinal tract: “Juvenile hamartomatous polyps”
Ciliary Body Medulloepithelioma
 Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
CBMEs in DICER1 syndrome have occurred in children under 10 years of age. Fewer than 20 cases connected to DICER1 have been reported in the medical literature, so it is very unusual even in DICER1 syndrome
How is CBME diagnosed?
Because this tumor occurs at the front of the eye, it is sometimes noticed by parents as an unusual appearance of the pupil of the eye. Also, problems may be noticed in a school vision test. Vision in one eye may be somewhat or significantly impaired.
An eye doctor will easily notice an abnormality on a regular eye examination, and CT and/or MRI scans will detect changes in the front of the eyeball. Generally, a very highly specialized eye doctor will be consulted who will recognize the eye abnormality as a possible CBME.
How is CBME treated?
Any eye tumor in a child requires the attention of very highly specialized eye physicians. Depending on the size of the tumor and the degree of impaired vision, the eye may have to be removed. However, sometimes, the eye can be saved despite some reduced vision.
When to test for DICER1 mutations:
CBME is a very unusual tumor and appears to occur with excess frequency in individuals with DICER1 mutations. CBME also occurs in individuals without DICER1 mutations. Thus, if a child is diagnosed with CBME, it is reasonable to inquire carefully within the child’s family to learn whether any other DICER1-mutation-associated illnesses are known. If such illnesses exist, then DICER1 testing of the child with CBME (or of the individuals with other conditions) is very reasonable. If no other DICER1-associated illnesses are noted in the family, it is less compelling to test for DICER1 mutation, but still reasonable to do so if the family wishes.
Nasal Chondromesenchymal hamartoma
 Like many tumors associated with DICER1 mutations, nasal chondromesenchymal hamartoma (NCMH) is very unusual, yet it occurs sometimes in DICER1 syndrome. It occurs in the nasal cavity and the sinuses adjacent to the nasal cavity. (Sinuses are essentially air-filled cul-de-sacs, or “out-croppings”, attached to the nasal cavity. Sinuses make various facial bones lighter in weight by creating air-filled pockets within them.)
Like many tumors associated with DICER1 mutations, nasal chondromesenchymal hamartoma (NCMH) is very unusual, yet it occurs sometimes in DICER1 syndrome. It occurs in the nasal cavity and the sinuses adjacent to the nasal cavity. (Sinuses are essentially air-filled cul-de-sacs, or “out-croppings”, attached to the nasal cavity. Sinuses make various facial bones lighter in weight by creating air-filled pockets within them.)
NCMH is a benign tumor – a benign growth. It is named by pathologists for its appearance under the microscope: “chondromesenchymal” means that the tissue has features of cartilage and other connective tissues. The word hamartoma is quite difficult to define but generally means that the tumor material looks fairly normal, but there is too much of it.
NCMH may be bilateral – that is, affect right and left sinuses at the same time. As an NCMH tumor enlarges, it can push aside and distort small facial bones, but it pushes them (instead of invading them), which is consistent with the concept that NCMH is not malignant.
How is NCMH diagnosed?
In DICER1 syndrome, NCMH has occurred in young people between the ages of approximately 7 to 20 years. Fewer than 20 cases connected to DICER1 have been reported in the medical literature, so it is very unusual even in DICER1 syndrome. In the general population, NCMH is also very unusual, and the diagnosis may be made at any age, with a preponderance of cases in young children under 2 years of age. Individuals with NCMH have nasal congestion and “stuffiness” as a sign of partially-blocked or blocked nasal passages. Physicians may notice unusual tissue in the nasal cavity. A CT or MRI scan reveals soft tissue filling the nasal cavity and/or adjacent sinuses. Surgical biopsy is necessary for diagnosis.
How is NCMH treated?
Surgical removal is generally successful for NCMH. Rarely, more than one surgery may be needed to remove all NCMH or recurrent NCMH (which is unusual).
When to test for DICER1 mutations:
NCMH, like CBME discussed above, is very unusual both in the general population and in DICER1 syndrome. Based on current information, it appears that individuals with a DICER1 mutation have a slightly increased chance of developing NCMH. NCMH also occurs with no connection to DICER1 syndrome. Thus, if a child is diagnosed with NCMH, it is reasonable to inquire carefully within the child’s family to learn whether any other DICER1-syndrome-associated illnesses are known. If such illnesses exist, then DICER1 testing of the child with NCMH (or of the individuals with other conditions) is very reasonable. If no other DICER1-associated illnesses are noted in the family, it is less compelling to test for DICER1 mutations, but still reasonable to do so if the family wishes.
Juvenile Intestinal Polyps
 Juvenile intestinal polyps are small grape size lumps or clusters growing from the inside surface of the intestinal tube. Although named ‘juvenile’ polyps because of a specific appearance under the microscope, these polyps tend to occur not only in early childhood but up to the ages of 15-20 years. They may partially block the intestine, slowing the passage of intestinal contents. They are benign. There is likely to be only one or a few polyps. They occur in the general population as well as in DICER1 syndrome. They are unusual in DICER1 syndrome, and although proof does not yet exist that they are directly connected to DICER1 mutation, it appears as if there is a connection.
Juvenile intestinal polyps are small grape size lumps or clusters growing from the inside surface of the intestinal tube. Although named ‘juvenile’ polyps because of a specific appearance under the microscope, these polyps tend to occur not only in early childhood but up to the ages of 15-20 years. They may partially block the intestine, slowing the passage of intestinal contents. They are benign. There is likely to be only one or a few polyps. They occur in the general population as well as in DICER1 syndrome. They are unusual in DICER1 syndrome, and although proof does not yet exist that they are directly connected to DICER1 mutation, it appears as if there is a connection.
How are juvenile intestinal polyps diagnosed?
In DICER1 syndrome, these polyps tend to occur in the small intestine and they eventually block/obstruct the intestinal tract causing pain and bloating. Various x-ray techniques such as CT scan or barium enema (or a similar technique for young children) will identify a blockage and may show the polyps inside the intestinal tube. When a polyp occurs elsewhere in the intestinal tract (such as in the esophagus or rectum, which occurs rarely), other diagnostic approaches will be used.
How are juvenile intestinal polyps treated?
Surgery is used to remove the polyp or polyps and relieve any intestinal blockage.
When to test for DICER1 mutations:
Juvenile intestinal polyps are unusual in DICER1 syndrome and probably are somewhat more frequent in the general population than in DICER1 mutation carriers. If an individual is diagnosed with juvenile intestinal-tract polyps and there are no other DICER1-related conditions in the individual or family, DICER1 mutation testing does not appear to be indicated. If juvenile polyps occur with any other DICER1 condition, testing is reasonable.

 A few unusual brain tumors occur in DICER1 syndrome. One brain tumor, pituitary blastoma, is particularly characteristic of DICER1 syndrome, and is discussed here.
A few unusual brain tumors occur in DICER1 syndrome. One brain tumor, pituitary blastoma, is particularly characteristic of DICER1 syndrome, and is discussed here.
 One tumor of the ovary (Sertoli-Leydig cell tumor) is among the frequent manifestations of DICER1 mutation in females. Another ovarian tumor and a tumor of the uterine cervix (called embryonal rhabdomyosarcoma) are very rare manifestations of DICER1 mutation.
One tumor of the ovary (Sertoli-Leydig cell tumor) is among the frequent manifestations of DICER1 mutation in females. Another ovarian tumor and a tumor of the uterine cervix (called embryonal rhabdomyosarcoma) are very rare manifestations of DICER1 mutation.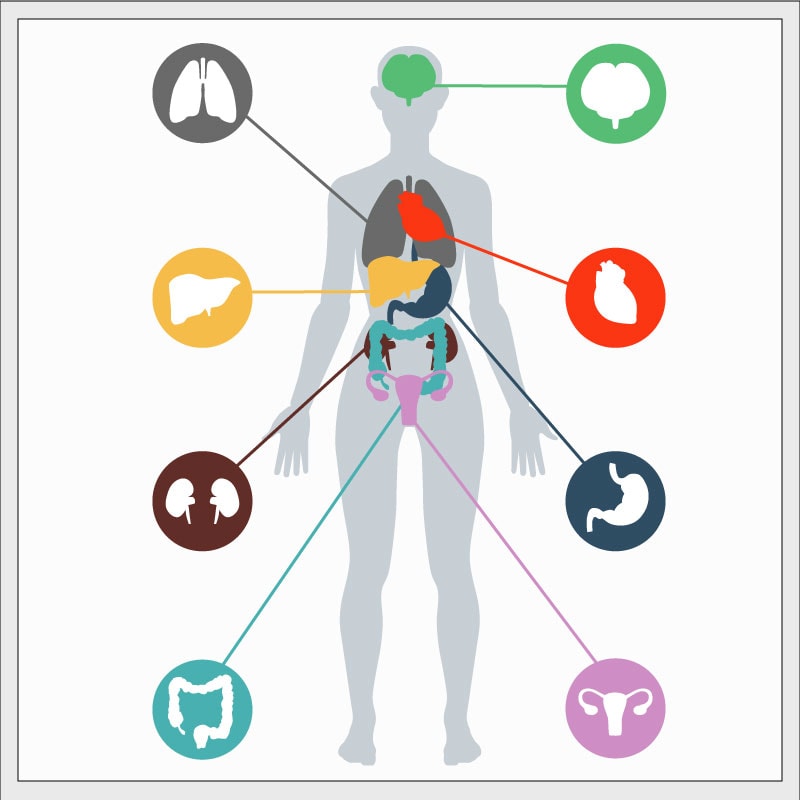 One of the most distinctive overall features of DICER1 syndrome is that mutation carriers are susceptible to very unusual conditions. Although these conditions are rare in general, they nevertheless occur in this syndrome with what appears to be increased frequency. Though they seem to occur more frequently in DICER1 syndrome than in the general population, these conditions remain unusual in the syndrome.
One of the most distinctive overall features of DICER1 syndrome is that mutation carriers are susceptible to very unusual conditions. Although these conditions are rare in general, they nevertheless occur in this syndrome with what appears to be increased frequency. Though they seem to occur more frequently in DICER1 syndrome than in the general population, these conditions remain unusual in the syndrome.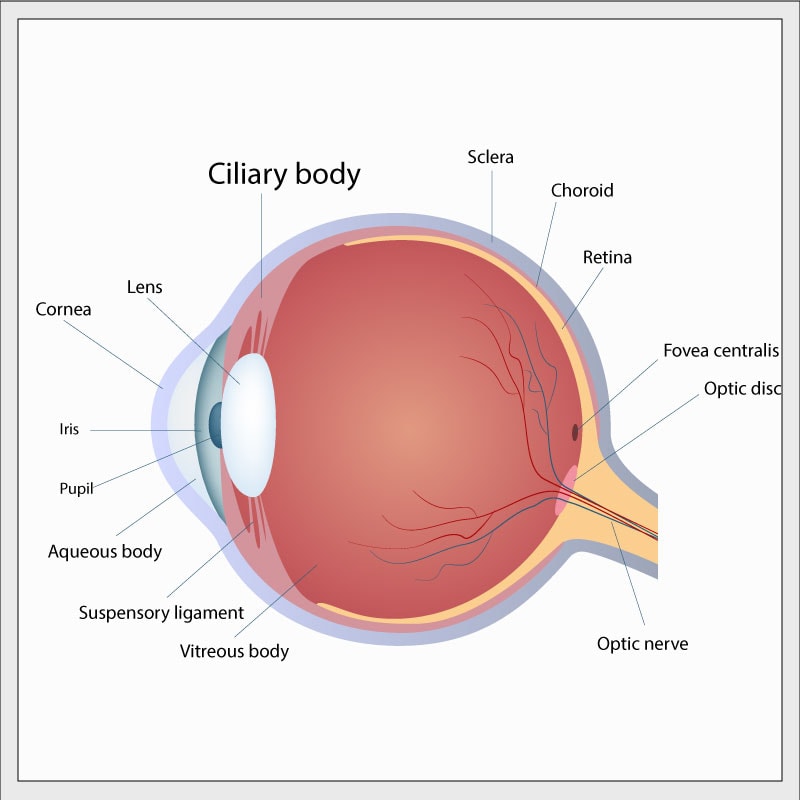 Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign.
Ciliary body medulloepithelioma (CBME) is a small tumor in the front of the eye. The “ciliary body” is a tiny, complex ring-shaped structure inside and at the front of the eyeball surrounding the lens. Muscles in the ciliary body control the shape of the lens and other parts of the ciliary body carry blood vessels to supply and control the inner contents of the eye. “Medulloepithelioma” is a very complex pathologist’s term to describe the microscopic appearance of certain kinds of abnormal tissue. CBME can be malignant or benign – to date, the very few CBME which have been associated with DICER1 mutation have been benign. Like many tumors associated with DICER1 mutations, nasal chondromesenchymal hamartoma (NCMH) is very unusual, yet it occurs sometimes in DICER1 syndrome. It occurs in the nasal cavity and the sinuses adjacent to the nasal cavity. (Sinuses are essentially air-filled cul-de-sacs, or “out-croppings”, attached to the nasal cavity. Sinuses make various facial bones lighter in weight by creating air-filled pockets within them.)
Like many tumors associated with DICER1 mutations, nasal chondromesenchymal hamartoma (NCMH) is very unusual, yet it occurs sometimes in DICER1 syndrome. It occurs in the nasal cavity and the sinuses adjacent to the nasal cavity. (Sinuses are essentially air-filled cul-de-sacs, or “out-croppings”, attached to the nasal cavity. Sinuses make various facial bones lighter in weight by creating air-filled pockets within them.) Juvenile intestinal polyps are small grape size lumps or clusters growing from the inside surface of the intestinal tube. Although named ‘juvenile’ polyps because of a specific appearance under the microscope, these polyps tend to occur not only in early childhood but up to the ages of 15-20 years. They may partially block the intestine, slowing the passage of intestinal contents. They are benign. There is likely to be only one or a few polyps. They occur in the general population as well as in DICER1 syndrome. They are unusual in DICER1 syndrome, and although proof does not yet exist that they are directly connected to DICER1 mutation, it appears as if there is a connection.
Juvenile intestinal polyps are small grape size lumps or clusters growing from the inside surface of the intestinal tube. Although named ‘juvenile’ polyps because of a specific appearance under the microscope, these polyps tend to occur not only in early childhood but up to the ages of 15-20 years. They may partially block the intestine, slowing the passage of intestinal contents. They are benign. There is likely to be only one or a few polyps. They occur in the general population as well as in DICER1 syndrome. They are unusual in DICER1 syndrome, and although proof does not yet exist that they are directly connected to DICER1 mutation, it appears as if there is a connection.