Syndrome DICER1 : Génétique, hérédité et principales caractéristiques cliniques

Le syndrome DICER1 est un syndrome de prédisposition tumorale familiale qui découle de mutations génétiques touchant un gène appelé DICER1. Certaines des tumeurs liées au syndrome DICER1 sont très graves et présentent un danger de mort, mais un grand nombre des tumeurs qui y sont liées sont loin d’être aussi graves. En général, les tumeurs sont rares ou très rares, et certaines sont si rares qu’elles peuvent survenir uniquement chez des personnes présentant une mutation de DICER1.
Il ne semble exister aucune tendance particulière en ce qui a trait à la formation des tumeurs, sauf que chaque tumeur liée au syndrome DICER1 tend à apparaître à certains âges particuliers. Chez les personnes atteintes de ce syndrome, les tumeurs surviennent habituellement au cours de l’enfance, et même au cours de la première enfance, ou avant l’âge de 30 ans. On estime que plus de la moitié des personnes susceptibles d’être atteintes du syndrome DICER1 n’éprouve aucun problème médical.
Qu’est-ce que le gène DICER1, et que fait-il?
En termes simples, le gène DICER1 indique à chaque cellule humaine les autres instructions génétiques qu’elle doit suivre. Plus précisément, le gène DICER1 ordonne à la cellule de produire une protéine constituant un important mécanisme de contrôle des activités cellulaires. Presque tous les organismes vivants ont besoin de ce genre de contrôle et, par conséquent, la plupart des organismes vivants ont des gènes Dicer. « DICER1 » est le nom précis de la version humaine du gène Dicer, qui a en fait d’abord été découvert dans un petit ver!
Modes de transmission
Les êtres humains possèdent deux copies du gène DICER1 dans chaque cellule de leur corps. Habituellement, la prédisposition au syndrome DICER1 découle de la transmission d’un exemplaire anormal (« mutant ») du gène DICER1. Étant donné que les cellules comportent deux exemplaires de chaque gène, les cellules fonctionnent généralement de façon acceptable même lorsqu’un des exemplaires est anormal. Le gène mutant est transmis par la mère ou le père du patient. Le mode de transmission du syndrome DICER1 est désigné par l’appellation scientifique transmission « autosomique dominante ». L’un des parents porte un exemplaire normal et un exemplaire mutant du gène DICER1. Un enfant de ce parent héritera de l’exemplaire normal ou de l’exemplaire anormal; les chances de transmission de l’une ou l’autre de ces versions sont de 50 %. Chacun des enfants de ce parent a une chance sur deux d’en hériter. Il y a de fortes chances que la moitié des enfants de ce parent hériteront de l’exemplaire normal et que l’autre moitié héritera de l’exemplaire mutant, et seuls ces derniers seront susceptibles d’être atteints de maladies liées au syndrome DICER1. Une telle mutation est présente dans toutes les cellules de la personne et peut notamment être décelée dans les cheveux, la salive, et le sang. On estime que seulement 1 bébé sur 10 000 porte des mutations de DICER1 à la naissance; il serait donc extrêmement rare que les deux parents portent une mutation de DICER1, et aucun cas d’enfant ayant des mutations de DICER1 provenant des deux parents n’a jamais été observé.
Dans probablement 10 à 15 % des cas, une mutation de DICER1 n’est pas héritée d’un parent. Il s’agit plutôt d’une nouvelle mutation qui survient chez l’enfant; on ne sait pas si la mutation s’est produite dans un spermatozoïde, dans l’ovule de la mère, ou très peu de temps après la conception. Une personne présentant une nouvelle mutation peut transmettre celle ci à ses enfants.
Il est possible mais extrêmement rare qu’une mutation de DICER1 soit présente uniquement dans certains tissus organiques de la personne touchée, par exemple dans les cellules pulmonaires ou dans les cellules rénales ou dans tout autre tissu particulier. Les questions à savoir comment ces mutations surviennent ou pourquoi elles ne touchent que certains tissus demeurent sans réponse. Seuls les tissus portant la mutation sont susceptibles d’être touchés par des affections liées à DICER1. On parle alors de mutations « en mosaïque » parce qu’elles apparaissent seulement dans certaines cellules de l’organisme.
Quels sont les signes qu’une personne puisse être porteuse d’une mutation dans DICER1?
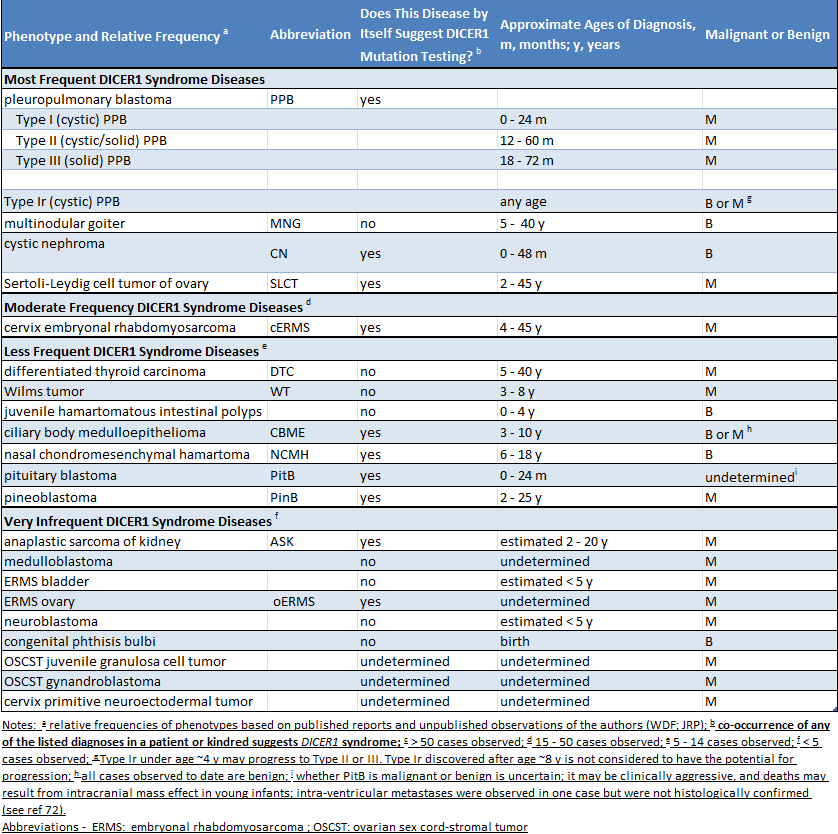
Étant donné qu’un grand nombre de personnes présentant une mutation de DICER1 n’ont aucun problème médical, il se peut très bien qu’un parent ignore qu’il est porteur d’une mutation. La présence d’une mutation est soupçonnée dans deux situations. Premièrement, lorsqu’un enfant reçoit un diagnostic d’une maladie très étroitement liée au syndrome DICER1 (comme un blastome pleuropulmonaire), il convient de rechercher une mutation de DICER1. Deuxièmement, si deux ou plusieurs des maladies liées au syndrome DICER1 sont diagnostiquées chez un enfant ou chez ses frères ou sœurs ou d’autres parents proches, il est raisonnable de rechercher une mutation de DICER1. Le tableau suivant présente une liste des maladies liées au syndrome DICER1. Le tableau indique si les affections énumérées sont surtout associées au syndrome DICER1 (colonne 3, « oui ») ou si elles surviennent au sein de la population générale et sont également plus rarement liées au syndrome DICER1 (colonne 3, « non »)
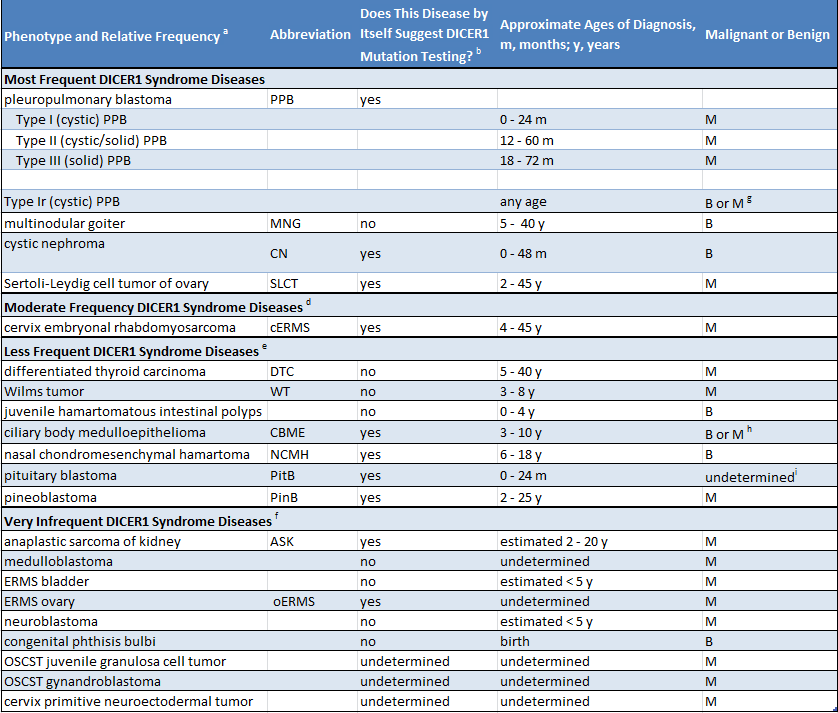
(Clicker pour agrandir)
Voici des exemples de cas dans lesquels il faut soupçonner la présence du syndrome DICER1. Lorsqu’un enfant reçoit un diagnostic de blastome pleuropulmonaire (BPP) ou de blastome de l’hypophyse, il y a lieu de soupçonner fortement qu’il est atteint du syndrome DICER1, car approximativement 75 % des patients présentant un BPP portent une mutation de DICER1 et plus de 90 % des patients ayant un blastome de l’hypophyse ont une telle mutation. En revanche, par exemple, si un enfant reçoit un diagnostic de tumeur de Wilms au rein, les soupçons sont peu élevés parce que seul un très petit pourcentage de patients ayant une tumeur de Wilms (moins de 5 %) ont une mutation de DICER1. Certaines combinaisons de maladies (chez une personne donnée ou des membres de sa famille proche) font fortement soupçonner la présence du syndrome DICER1 : par exemple, probablement environ 50 % des tumeurs à cellules de Sertoli-Leydig de l’ovaire sont causées par des mutations de DICER1, mais lorsque des tumeurs touchent aussi la thyroïde et s’accompagne de nodules (goitre multinodulaire) chez le patient ou des membres de sa famille, cette combinaison laisse fortement entendre qu’une mutation de DICER1 est à l’origine des deux affections.
Mutations de DICER1 dans les tumeurs
En dernier lieu, le présent exposé sur la génétique de DICER1 doit inclure un fait concernant les gènes DICER1 se trouvant dans spécifiquement dans les tumeurs. Des scientifiques ont déterminé que les tumeurs mêmes présentent habituellement deux mutations, une dans chacun des deux exemplaires du gène DICER1. La première mutation est la « mutation constitutionnelle » de la personne (elle est présente sur une copie du gène dans toutes les cellules). La deuxième mutation touche le second exemplaire du gène et est présente uniquement dans les cellules tumorales. On ne sait pas comment, pourquoi ou quand la deuxième mutation se produit. Toutefois, on croit que la tumeur s’est formée parce qu’aucun des exemplaires du gène n’était normal. Même les maladies bénignes liées à DICER1 présentent deux exemplaires mutants du gène.
Blastome pleuropulmonaire

Le blastome pleuropulmonaire (BPP) est une tumeur maligne du poumon qui est rare et survient durant la petite enfance. Elle a été reconnue pour la première fois par des médecins aux environs de 1985. Bien entendu, le BPP existait avant 1985, mais il était si rare que les médecins ne l’avaient jamais reconnu comme une maladie distincte. Le BPP diffère complètement du cancer du poumon touchant les adultes et n’a aucun lien avec celui ci. Étant donné que cette maladie rare, le BPP, survenait dans certaines familles, la recherche d’une cause a mené à la découverte des mutations de DICER1, dont on sait maintenant qu’elles causent la grande majorité des cas de BPP.
Quand survient-il?
Le BPP survient presque exclusivement chez des enfants âgés de moins de quatre ans. Il peut aussi toucher les bébés. La plupart des enfants atteints de BPP présentent une mutation de DICER1, mais de rares cas se produisent en l’absence de cette mutation. Le BPP touche très rarement les enfants plus âgés et les adultes.
Quels sont les types de BPP?
Le BPP se manifeste sous trois formes principales : premièrement, chez les enfants de moins d’un an, le BPP apparaît sous forme de « kystes » dans les poumons – les kystes de BPP sont des cavités remplies d’air qui résultent de la croissance anormale des poumons. Ces kystes peuvent être petits et ne provoquer aucun symptôme, tandis que d’autres peuvent être très gros et entraîner des difficultés respiratoires légères ou graves. Le BPP kystique est appelé BPP de type I.
La plupart des kystes du poumon survenant chez les bébés ne sont PAS des BPP; il s’agit d’une affection complètement différente et moins grave qui n’a rien à voir avec les mutations de DICER1. La chirurgie permet de supprimer les kystes sans autres problèmes. Étant donné que le BPP est rare et que d’autres kystes sont plus fréquents, les médecins considèrent presque toujours que les kystes du poumon appartiennent au type le plus courant. Le diagnostic de BPP peut être posé seulement après que le kyste a été retiré puis examiné au microscope. Les radiographies du BPP et des kystes les plus courants sont semblables. L’ablation des kystes de BPP constitue habituellement un traitement curatif.
La deuxième forme de BPP (BPP de type II) survient généralement chez les enfants âgés de 12 à 30 mois. Ces BPP se composent à la fois de kystes et de masses tumorales solides. La troisième forme de BPP se compose de tumeurs solides sans kystes – le BPP de type III – et survient habituellement chez les enfants âgés de 2 à 4 ans. Les tumeurs de BPP de types II et III peuvent entraîner des difficultés respiratoires, des signes généraux de maladie ou une fièvre, et se présentent sous forme d’« ombres » sur une radiographie des poumons; étant donné que le BPP est rare par comparaison à la pneumonie, il arrive couramment que les médecins pensent initialement qu’un enfant a une pneumonie. Les BPP de types II et III sont graves et malins et exigent le recours à la chirurgie et à la chimiothérapie et parfois à la radiothérapie. De nombreux enfants guérissent de ces tumeurs, mais malheureusement, d’autres peuvent mourir de ces formes de BPP.
Comme il est mentionné ci dessus, les plages d’âge des personnes touchées par les BPP de types I, II, et III ne sont pas précises. En outre, le BPP de type I peut se transformer en BPP de type II ou III et par conséquent, tous ces types sont reliés et représentent un « éventail » de manifestations du BPP. Il existe une forme très rare de BPP de type I, appelée BPP de type Ir, que l’on croit être une forme non maligne de BPP de type I et qui ne se transforme pas en BPP de type II ou III.
Quand tester pour des mutations dans DICER1?
Le BPP est très caractéristique de la mutation de DICER1. Il est donc raisonnable de procéder au dépistage de mutations de DICER1 chez un enfant ayant reçu un diagnostic de BPP.
Thyroid Disease in DICER1 Syndrome
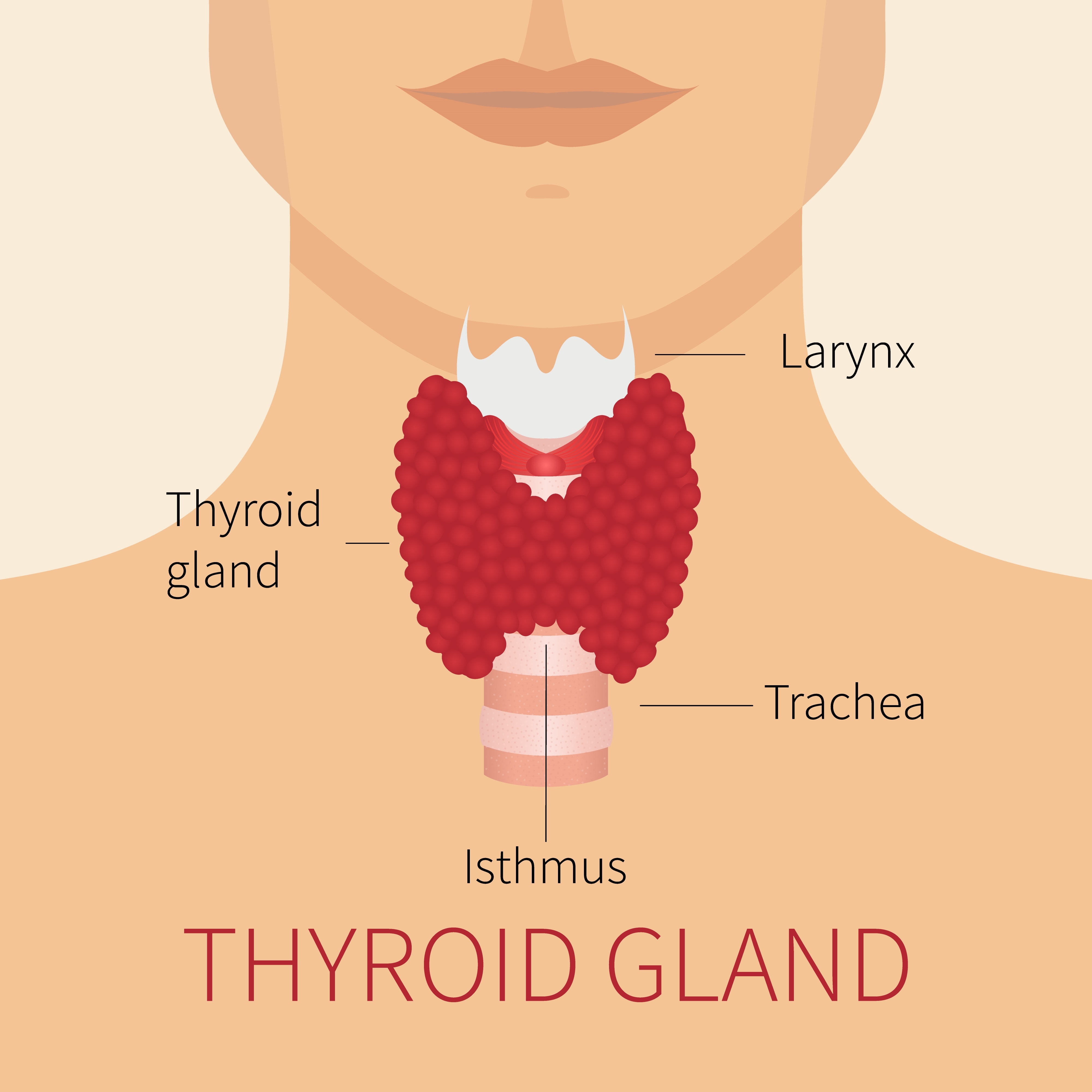
La glande thyroïde est le site des anomalies le plus fréquemment associées aux mutations de DICER1. La glande est située à l’avant du cou, juste en dessous du larynx (« pomme d’Adam ») et comporte des lobes en forme d’aile de papillon de chaque côté de la trachée, comme l’indique le diagramme adjacent.
La glande thyroïde produit une hormone qui contrôle de nombreuses actions dans le corps, ce que l’on peut décrire de façon générale comme le réglage du thermostat énergétique du corps. Lorsque la quantité d’hormone thyroïdienne est trop élevée, la personne touchée perd du poids trop rapidement et a tendance à avoir trop chaud; lorsque la quantité d’hormone thyroïdienne est trop faible, le corps fonctionne au ralenti et la personne a froid, prend du poids et souffre de constipation. Les affections de la glande thyroïde présentes dans le cadre du syndrome DICER1, qui sont décrites ci dessous, ne modifient pas l’activité de l’hormone thyroïdienne.
Dans le cadre du syndrome DICER1, la fonction de contrôle hormonal de la glande n’est pas touchée, mais des masses (tumeurs) bénignes peuvent se former dans la glande et la faire grossir en faisant apparaître un gonflement à la partie inférieure avant du cou, appelé « goitre ». Ces masses sont rarement malignes.
Goitre multinodulaire
L’anomalie la plus fréquente dans le cadre du syndrome DICER1 est l’apparition de nombreuses masses bénignes (nodules et kystes) réparties dans toute la glande. Lorsqu’elles sont suffisamment grosses pour être visibles, elles forment ce que l’on appelle un « goitre multinodulaire ». Les deux côtés de la glande sont habituellement touchés. Les nodules ne sont pas malins. L’activité hormonale n’est pas modifiée. Ces goitres sont beaucoup plus fréquents chez les porteurs de mutations de sexe féminin que chez ceux de sexe masculin. Selon les estimations, une grande proportion de porteurs de mutations de sexe féminin et une proportion très inférieure de porteurs de sexe masculin présenteront un goitre multinodulaire au cours de leur vie.
Cette affection – le goitre multinodulaire (GMN) – est très fréquente au sein de la population générale. Par conséquent, le GMN en soi n’évoque pas la présence de mutations de DICER1 ou du syndrome DICER1. Toutefois, le GMN apparaît habituellement à un âge plus précoce chez les porteurs de mutations que chez les membres de la population générale. Chez les porteurs de mutations de DICER1, le GMN apparaît généralement entre l’âge de 10 et 30 ans, mais il peut se produire dès l’âge de 5 ans.
Carcinome différencié de la thyroïde
Le cancer différencié de la thyroïde (ou « carcinome différencié de la thyroïde ») (CDT) survient rarement dans le cadre du syndrome DICER1. Dans le cas de cette affection, des masses se forment dans la thyroïde, comme dans le cas du GMN décrit ci dessus, mais le tissu est malin plutôt que bénin. Le terme « différencié » est utilisé pour indiquer que le tissu malin n’a pas une apparence primitive; le tissu primitif peut indiquer la présence d’une tumeur maligne agressive. Le CDT n’est pas un cancer agressif. Il s’agit habituellement d’un cancer de « bas grade », ce qui signifie qu’il reste généralement confiné à la glande thyroïde et peut être guéri au moyen d’une intervention chirurgicale.
Comment sont ces conditions diagnostiquées?
D’ordinaire, le GMN et le CDT se présentent sous forme de renflement à la base du cou (« goitre »). Grâce à un examen physique minutieux, les médecins peuvent déceler des masses dans la thyroïde (GMN ou CDT) avant qu’elles deviennent perceptibles sous forme de goitre. Un examen par échographie est un test extrêmement fiable et non invasif qui permet de définir toute anomalie de la glande thyroïde et peut révéler des différences entre le GMN et le CDT. Il est nécessaire d’effectuer des biopsies à l’aiguille pour déterminer la nature réelle des nodules de la thyroïde
Comment sont ces conditions traitées?
Le GMN peut être observé sans faire l’objet d’un traitement particulier si les nodules sont petits et ne grossissent pas. Si la glande thyroïde grossit en raison de la présence de nombreux nodules de GMN ou de CDT, on procède à son ablation chirurgicale. L’administration quotidienne de médicaments permet très bien de remplacer l’hormone thyroïdienne..
Quand tester pour des mutations dans DICER1?
Le GMN et le CDT sont caractéristiques de la mutation de DICER1, mais sont également relativement fréquents au sein de la population générale. Par conséquent, aucun de ces diagnostics en soi ne justifie le dépistage de mutations.
Par contre, lorsqu’un GMN ou un CDT et toute autre maladie liée au syndrome DICER1 surviennent chez une personne ou chez les membres de sa famille proche, il est raisonnable de procéder au dépistage de mutations de DICER1. La présence simultanée d’un GMN et d’une tumeur à cellules de Sertoli-Leydig de l’ovaire semble particulièrement indiquer l’existence d’une mutation de DICER1.
Blastome de l’hypophyse dans le cadre du syndrome DICER1
 Quelques tumeurs inhabituelles du cerveau se forment dans le cadre du syndrome DICER1. Nous abordons plusieurs de celles ci dans une autre section. Une tumeur du cerveau, le blastome de l’hypophyse, est très inhabituelle et est particulièrement caractéristique du syndrome DICER1; nous l’abordons ici.
Quelques tumeurs inhabituelles du cerveau se forment dans le cadre du syndrome DICER1. Nous abordons plusieurs de celles ci dans une autre section. Une tumeur du cerveau, le blastome de l’hypophyse, est très inhabituelle et est particulièrement caractéristique du syndrome DICER1; nous l’abordons ici.
Qu’est-ce que l’hypophyse?
L’hypophyse est une petite glande se trouvant à la face inférieure du cerveau au centre de la tête et juste derrière la paroi arrière des voies nasales. L’hypophyse envoie dans la circulation sanguine des hormones de contrôle central qui, à leur tour, contrôlent plusieurs autres organes produisant des hormones, notamment la thyroïde, les ovaires, les testicules, et les glandes surrénales.
Qu’est-ce que le blastome de l’hypophyse??
Le blastome de l’hypophyse est une tumeur de l’hypophyse qui semble être presque uniquement liée au syndrome DICER1. Il est extrêmement rare et a été décrit dans la littérature médicale seulement en 2008 et a été lié aux mutations de DICER1 en 2014. Moins de 20 cas ont été reconnus et signalés dans la littérature médicale à l’échelle mondiale. Le terme « blastome » est utilisé par les pathologistes qui décèlent au microscope une sorte particulière de tissu qui ressemble à celui d’un organe particulier dans un embryon humain précoce.
On ne sait pas si le blastome de l’hypophyse est malin ou bénin, mais étant donné qu’il se situe au centre de la tête et touche de jeunes enfants, il s’agit d’une tumeur grave.
Le blastome de l’hypophyse survient chez de très jeunes enfants et le plus ancien cas reconnu touchait un enfant de 2 ans. La tumeur peut faire grossir l’hypophyse et peut faire saillie au dessus de l’hypophyse.
Quels sont les symptômes du blastome de l’hypophyse?
Le plus fréquemment, le blastome de l’hypophyse semble causer un dérèglement hormonal qui provoque un accroissement des hormones surrénales, ce qui entraîne une augmentation de l’appétit et un gain de poids selon un schéma particulier appelé maladie de Cushing – le genre de gain de poids observé chez les personnes qui doivent utiliser des « stéroïdes » à des fins de traitement médical. Les enfants touchés prennent du poids lentement au cours de plusieurs mois, ce qui donne à penser que la tumeur croît lentement. Le blastome de l’hypophyse peut aussi provoquer une incoordination des mouvements des yeux. Parmi les divers autres symptômes possibles, mentionnons notamment la fatigue, le ralentissement de la croissance staturale, et la perturbation des voies de circulation des fluides dans le cerveau (« hydrocéphalie »).
Comment diagnostiquer et traiter le blastome de l’hypophyse?
Les symptômes du blastome de l’hypophyse sont habituellement très marqués, ce qui incite les médecins à les étudier de façon intensive. L’extrême rareté du blastome de l’hypophyse fait en sorte que les médecins envisagent rarement la possibilité de sa présence, et ce, jusqu’à ce que l’étude soit très avancée. Un tomodensitogramme ou un examen par imagerie par résonance magnétique (IRM) de la tête permet de déceler rapidement une tumeur. La chirurgie peut permettre de retirer la tumeur ou d’en réduire considérablement la taille. On ne sait pas si un traitement allant au delà de la chirurgie s’impose en ce qui concerne le blastome de l’hypophyse. Certaines personnes semblent avoir vécu plusieurs années et avoir été guéries. Les déséquilibres hormonaux sont traités au moyen de divers traitements hormonaux de substitution.
Quand tester pour des mutations dans DICER1?
Puisque le blastome de l’hypophyse est très caractéristique du syndrome DICER1, il est fortement indiqué de procéder au dépistage de mutations de DICER1 chez tout enfant présentant cette tumeur.
Kystes et tumeurs au rein dans le cadre du syndrome DICER1
Les reins sont les organes principaux du système urinaire. Ils servent plusieurs fonctions vitales incluant l’élimination des toxines du corps par voie de l’urine. Les kystes et les tumeurs des reins ont été observés chez bon nombre de patient souffrant du syndrome DICER1.
Comment le syndrome DICER1 affecte-t-il les reins?
Plusieurs affections rénales peuvent survenir à cause de mutations de DICER1. La plus fréquente est le « néphrome kystique », qui touche environ de 5 % à 7 % des enfants présentant une mutation de DICER1. De plus, deux tumeurs malignes distinctes peuvent se former.
Néphrome kystique
Le néphrome kystique (NK) est une anomalie bénigne qui peut toucher un rein ou les deux reins chez les enfants présentant une mutation de DICER1. Il s’agit de l’une des affections les plus fréquentes dans le cadre du syndrome DICER1. Le NK se manifeste dans les tissus réguliers du rein sous forme de kystes arrondis remplis de fluide qui ressemblent à un ballon. Ces kystes peuvent être petits et mesurer de 0,5 à 1 pouce de diamètre, ou se présenter sous forme de très grandes grappes de kystes qui peuvent atteindre plusieurs pouces de diamètre. Les kystes peuvent altérer les tissus normaux des reins et parfois les remplacer. Il arrive que l’on désigne le NK sous le nom de « tumeur » parce que du point de vue médical, une « tumeur » est une bosse ou une masse; toutefois, le NK n’est pas cancéreux ni malin.
Quels sont les symptômes du néphrome kystique, et comment est-il traité?
Le NK survient presque toujours chez les enfants entre la naissance et l’âge de 4 ans; il peut survenir plus tard, jusqu’à l’âge de 12 à 15 ans, mais cela est rare. Étant donné que les jeunes enfants ne peuvent expliquer avec exactitude ce qu’ils ressentent, on ne sait pas si le NK cause de la douleur, mais la douleur ne constitue probablement pas un problème important dans le cas du NK. Le NK est habituellement porté à l’attention des médecins parce que les parents remarquent la présence d’une « masse » ou d’une bosse à l’abdomen. Certains kystes du NK sont si petits qu’ils sont découverts de façon fortuite lorsqu’un enfant subit une radiographie ou un autre examen par imagerie pour d’autres raisons.
Le NK touche généralement un seul rein, mais il n’est pas particulièrement rare qu’il touche les deux reins. La chirurgie est utilisée comme traitement curatif du NK. On peut éliminer un NK de petite taille en retirant une petite partie du rein. Il est inhabituel que d’autres kystes se forment après l’enlèvement du NK. Si un NK de grande taille déforme un rein, il se peut que l’on retire le rein en entier. Malgré l’ablation d’un rein ou même d’un rein entier et d’une partie de l’autre rein, il est extrêmement rare que la fonction rénale d’un enfant soit compromise à la suite du traitement chirurgical du NK.
Si de très petits kystes rénaux sont découverts de façon fortuite chez un enfant présentant une mutation de DICER1, il s’agit fort probablement d’un NK. Le diagnostic précis ne peut être établi que si les kystes sont retirés puis examinés au microscope. Cependant, il se peut qu’il ne soit pas nécessaire de procéder à une ablation et d’établir un diagnostic précis lorsque les kystes sont petits et ne provoquent aucun symptôme. On ne connait pas à l’heure actuelle la meilleure méthode de traitement des petits kystes. Certes, certains médecins recommandent leur ablation, mais d’autres peuvent suggérer la réalisation d’un examen périodique par imagerie par des radiologues.
Y a-t-il des tumeurs malignes qui affectent les reins dans le contexte du syndrome DICER1?
Deux tumeurs malignes du rein peuvent survenir chez les porteurs de mutations de DICER1. Heureusement, les deux sont très inhabituelles.
Tumeur de Wilms
La « tumeur de Wilms » est une tumeur maligne du rein qui survient durant l’enfance – la plupart des cas se produisent avant l’âge de 10 ans et cette tumeur survient le plus souvent entre l’âge de 2 et 4 ans. La mutation de DICER1 n’est pas un facteur dans plus de 95 % des cas de tumeur de Wilms. Toutefois, très peu d’enfants portant une mutation de DICER1 présentent une tumeur de Wilms. Bien qu’elle soit maligne, la tumeur de Wilms est généralement très guérissable.
Sarcome anaplasique du rein
Le sarcome anaplasique du rein (SAR) est une tumeur du rein extrêmement rare, mais il semble être particulièrement lié à une mutation de DICER1 et au néphrome kystique (NK, abordé ci dessus). Ce n’est que depuis environ 2014 que le SAR a été reconnu comme étant associé au syndrome DICER1, et par conséquent, il reste beaucoup à apprendre au sujet de celui-ci. Le SAR peut être précédé de l’apparition d’un néphrome kystique, mais les interrelations entre le NK et le SAR ne sont toujours pas connues. Le NK est l’une des affections les plus fréquemment associées à une mutation de DICER1, tandis que le SAR est l’une des affections qui le sont le plus rarement. Il semble donc que le NK n’est que rarement suivi d’un SAR. Le SAR peut survenir avant l’âge de 2 ans, mais aussi jusqu’à l’âge de 20 ans et peut être au delà. Il est malin et semble exiger un traitement agressif, et des enfants ont certainement été guéris.
Quand tester pour des mutations dans DICER1?
Le NK et le SAR sont caractéristiques des mutations de DICER1. Par conséquent, si un enfant reçoit un diagnostic de NK ou de SAR, il est raisonnable de procéder au dépistage de mutations de DICER1. La tumeur de Wilms est si rarement associée aux mutations de DICER1 que le diagnostic de cette affection ne laisse pas entendre la présence d’une mutation de DICER1. Ce n’est que lorsqu’un patient présente une tumeur de Wilms ou lorsque d’autres membres de la famille sont atteints de l’une ou plusieurs des maladies associées au syndrome DICER1 (se reporter aux autres maladies abordées ici) que le dépistage de mutations est indiqué.
Tumeur à cellules de Sertoli-Leydig de l’ovaire et autres tumeurs touchant l’appareil reproducteur féminin dans le cadre du syndrome DICER1
 Une tumeur de l’ovaire (la tumeur à cellules de Sertoli-Leydig) compte parmi les manifestations fréquentes de la mutation de DICER1 chez les femmes. Une autre tumeur de l’ovaire et une tumeur du col de l’utérus (appelée rhabdomyosarcome embryonnaire) sont de très rares manifestations de la mutation de DICER1.
Une tumeur de l’ovaire (la tumeur à cellules de Sertoli-Leydig) compte parmi les manifestations fréquentes de la mutation de DICER1 chez les femmes. Une autre tumeur de l’ovaire et une tumeur du col de l’utérus (appelée rhabdomyosarcome embryonnaire) sont de très rares manifestations de la mutation de DICER1.
Qu’est-ce que la tumeur à cellules de Sertoli-Leydig?
La tumeur à cellules de Sertoli-Leydig (TCSL) de l’ovaire est l’une des manifestations les plus fréquentes du syndrome DICER1. Il s’agit d’une forme rare de tumeur de l’ovaire qui représente moins de 5 % de toutes les tumeurs de l’ovaire au sein de la population générale, mais la TCSL est très caractéristique du syndrome DICER1. On ne connait pas le pourcentage de femmes portant une mutation qui sont atteintes d’une TCSL, mais on estime que probablement moins de 10 % d’entre elles le sont. La TCSL est maligne, mais il ne s’agit généralement pas d’une tumeur maligne agressive, et la plupart des patientes guérissent à la suite de l’ablation de la tumeur.
Il n’existe aucun lien entre la TCSL de l’ovaire et les tumeurs malignes de l’ovaire plus courantes qui surviennent au sein de la population générale. Ces tumeurs de l’ovaire peuvent se former chez des femmes ayant une prédisposition au cancer du sein; encore une fois, il n’existe pas de lien entre la prédisposition au cancer du sein ou aux tumeurs de l’ovaire et le syndrome DICER1, et, selon les connaissances actuelles, les femmes portant une mutation de DICER1 ne présentent pas une prédisposition accrue au cancer du sein.
La TCSL fait partie d’une famille de tumeurs de l’ovaire appelées « tumeurs des cellules stromales ». D’autres tumeurs des cellules stromales surviennent très rarement dans le cadre du syndrome DICER1. Le « gynandroblastome » et la « tumeur juvénile de la granulosa » sont des exemples des tumeurs rares des cellules stromales qui apparaissent dans le cadre du syndrome DICER1.
Quand la tumeur à cellules de Sertoli-Leydig apparaît-elle, et quels sont les symptômes?
Dans le cadre du syndrome DICER1, la TCSL survient habituellement environ entre l’âge de 10 et 30 ans, et bien que cela arrive rarement, elle peut survenir chez des filles âgées d’à peine deux à cinq ans, ou jusqu’à l’âge de 50 ans chez les femmes.
Les symptômes de la TCSL peuvent être attribuables aux hormones que la TCSL peut produire, et comprennent une pilosité corporelle ou faciale accrue, l’approfondissement de la voix ou des changements menstruels. Il se peut qu’il n’y ait aucun changement hormonal, et que la tumeur soit décelée en raison de la présence d’un ballonnement abdominal (« une masse à l’abdomen ») ou de changements dans les habitudes intestinales et urinaires.
La TCSL de l’ovaire survenant dans le cadre du syndrome DICER1 peut toucher les deux ovaires dans de rares occasions. Lorsque cela se produit, la survenue de chaque tumeur constitue un événement distinct et ces tumeurs se forment habituellement quelques mois ou quelques années l’une après l’autre.
Qu’est-ce que le rhabdomyosarcome embryonnaire de l’ovaire?
Le rhabdomyosarcome embryonnaire (RMSE) de l’ovaire est une tumeur maligne très rare qui survient dans le cadre du syndrome DICER1. Il apparaît habituellement entre l’âge de 10 et 30 ans. Seuls quelques cas ont été reconnus. Le nom « rhabdomyosarcome embryonnaire » renvoie à une certaine apparence que présente la tumeur examinée au microscope. L’apparence que présente le RMSE est un élément commun de diverses tumeurs de DICER1 touchant différentes parties du corps.
Le RMSE de l’ovaire est porté à l’attention des médecins par la présence d’un ballonnement abdominal ou peut être par des changements menstruels. On procède à l’ablation de l’ovaire. Étant donné que peu de cas ont été signalés, il n’existe pas de modèle de soins établi, de sorte que les oncologues choisissent le traitement ultérieur en fonction de la situation particulière de la patiente.
Qu’est-ce que le rhabdomyosarcome embryonnaire du col de l’utérus?
Le RMSE peut aussi survenir sous forme de tumeur du col de l’utérus et est une manifestation très rare de la mutation de DICER1. Il apparaît habituellement entre l’âge de 10 et 20 ans et rarement par la suite, se présente sous forme de grappes de tumeurs dans le vagin et provoque de légers saignements qui sont facilement confondus avec des saignements menstruels anormaux. Lorsque des grappes de tumeurs sont présentes dans le vagin, le RMSE du col de l’utérus est parfois désigné sous le nom particulier de « sarcome botryoïde du col de l’utérus ».
Quand tester pour des mutations dans DICER1?
Le RMSE et certaines autres tumeurs des cellules stromales de l’ovaire survenant chez les filles et les jeunes femmes sont très caractéristiques de la mutation de DICER1, mais ils se produisent aussi au sein de la population générale. Si une femme reçoit un diagnostic de l’une de ces tumeurs, il est raisonnable de procéder au dépistage de la mutation de DICER1.
Le RMSE de l’ovaire et celui du col de l’utérus sont caractéristiques du syndrome DICER1 et touchent très rarement la population générale. Si une femme reçoit un diagnostic de l’une de ces tumeurs, il est raisonnable de procéder au dépistage de la mutation de DICER1.
Diverses tumeurs inhabituelles dans le cadre du syndrome DICER1

L’une des caractéristiques générales les plus distinctives du syndrome DICER1 tient au fait que les porteurs de la mutation sont susceptibles de présenter des affections très inhabituelles. Ces affections sont généralement rares, mais elles surviennent néanmoins à une fréquence apparemment accrue dans le cadre de ce syndrome. Bien qu’elles semblent se manifester plus fréquemment chez les personnes atteintes du syndrome DICER1 qu’au sein de la population générale, ces affections demeurent inhabituelles dans le cadre de ce syndrome.
En plus des tumeurs inhabituelles dont il est question dans d’autres sections, nous abordons ici trois autres affections rares :
- Tumeur de l’œil : « médulloépithéliome du corps ciliaire »
- Tumeur des sinus paranasaux : « hamartome chondromésenchymateux nasal »
- Polypes dans le tractus intestinal : « polypes hamartomateux juvéniles »
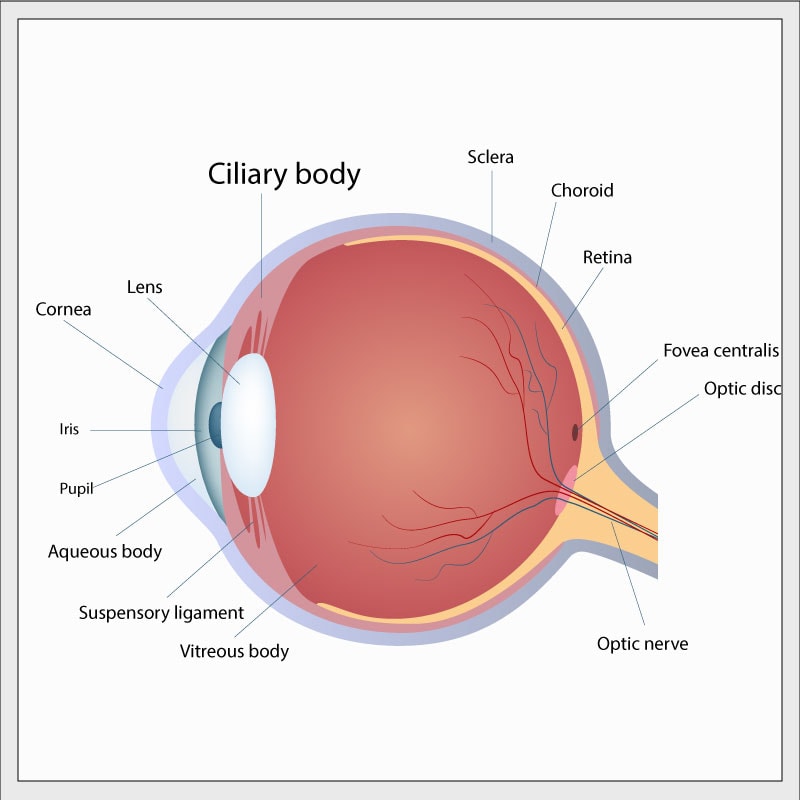
– Médulloépithéliome du corps ciliaire

Le médulloépithéliome du corps ciliaire (MECC) est une petite tumeur touchant la partie avant de l’œil. Le « corps ciliaire » est une minuscule structure complexe en forme d’anneau et se situe à la partie intérieure avant du globe oculaire autour de la lentille. Les muscles du corps ciliaire contrôlent la forme de la lentille, et d’autres parties du corps ciliaire portent les vaisseaux sanguins servant à alimenter et à contrôler les parties internes de l’œil. Le terme « médulloépithéliome » est très complexe et est utilisé par les pathologistes pour décrire l’apparence microscopique de certains types de tissus anormaux. Le MECC peut être malin ou bénin – à ce jour, les rares MECC qui ont été associés à la mutation de DICER1 étaient bénins.
Les MECC survenus dans le cadre du syndrome DICER1 touchaient des enfants âgés de moins d’une dizaine d’années. Moins de 20 cas liés à DICER1 ont été signalés dans la littérature médicale et, par conséquent, les MECC sont très inhabituels même dans le cadre de ce syndrome.
Comment est le MECC diagnostiqué?
Étant donné que cette tumeur se forme à la partie avant de l’œil, sa présence est parfois constatée par les parents, qui remarquent l’aspect inhabituel de la pupille de l’œil. En outre, des problèmes peuvent être décelés lors d’un examen de la vue effectué à l’école. Cette tumeur peut entraîner une déficience visuelle légère ou importante dans un œil.
Un ophtalmologiste remarquera facilement une anomalie lors d’un examen périodique de la vue, et le recours à un tomodensitogramme ou à un examen par IRM permet de déceler les changements survenant à la partie avant du globe oculaire. De façon générale, on consultera un ophtalmologiste hautement spécialisé qui reconnaîtra la possibilité que l’anomalie oculaire soit un MECC.
Comment est le MECC traité?
La présence d’une tumeur à l’œil chez un enfant exige l’attention d’ophtalmologistes hautement spécialisés. Selon la taille de la tumeur et l’ampleur de la déficience visuelle, il peut être nécessaire de procéder à l’ablation de l’œil. Il est cependant parfois possible de sauver l’œil, malgré une certaine diminution de la vision.
Quand tester pour des mutations dans DICER1?
Le MECC est une tumeur très inhabituelle et semble survenir à une fréquence excessive chez des personnes portant des mutations de DICER1. Le MECC survient également chez des personnes ne portant pas de mutations de DICER1. Par conséquent, si un enfant reçoit un diagnostic de MECC, il est raisonnable de s’informer soigneusement pour déterminer si d’autres maladies associées à une mutation de DICER1 ont été décelées au sein de la famille de celui ci. Si c’est le cas, il est très raisonnable de procéder au dépistage de mutations de DICER1 chez l’enfant présentant un MECC (ou chez les personnes atteintes d’autres maladies). Si aucune autre maladie associée à DICER1 n’est recensée au sein de la famille, il est moins nécessaire de procéder au dépistage de mutations de DICER1, mais il demeure raisonnable de le faire si la famille le souhaite.
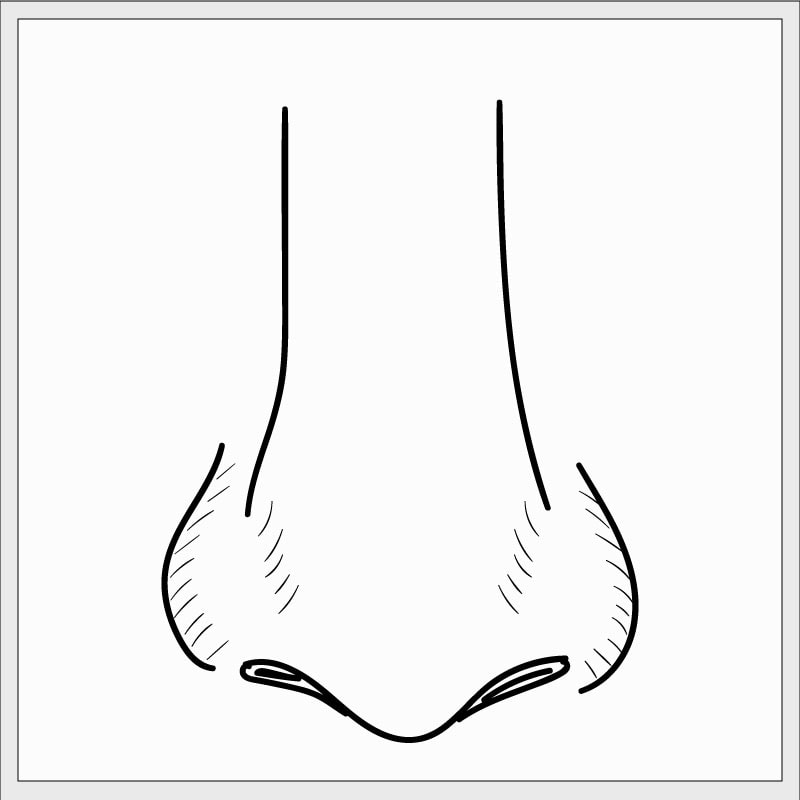
Hamartome chondromésenchymateux nasal

Comme un grand nombre des tumeurs associées aux mutations de DICER1, l’hamartome chondromésenchymateux nasal (HCMN) est très inhabituel, mais il survient parfois dans le cadre du syndrome DICER1. Il se forme dans la cavité nasale et dans les sinus adjacents à la cavité nasale. (Les sinus sont essentiellement des culs-de-sac remplis d’air, ou des « affleurements », rattachés à la cavité nasale. Les sinus allègent le poids de divers os du visage, car ils créent des poches remplies d’air à l’intérieur de ceux ci.)
Le HCMN est une tumeur bénigne – une masse bénigne. Son nom lui est attribué par les pathologistes en raison de l’apparence qu’il présente lors d’un examen au microscope : le terme « chondromésenchymateux » signifie qu’il comporte des caractéristiques du cartilage et d’autres tissus conjonctifs. Bien que le terme « hamartome » soit très difficile à définir, il signifie généralement que la matière tumorale a une apparence assez normale, mais qu’elle est trop abondante.
Le HCMN peut être bilatéral – c’est à dire qu’il touche simultanément les sinus droits et gauches. À mesure que la tumeur du HCMN grossit, elle peut écarter et déformer les petits os du visage, mais elle les repousse (au lieu de les envahir), ce qui cadre avec le concept selon lequel le HCMN n’est pas malin.
Comment est le HCMN diagnostiqué?
Dans le cadre du syndrome DICER1, le HCMN est survenu chez des jeunes âgés d’environ 7 à 20 ans. Moins de 20 cas liés à DICER1 ont été signalés dans la littérature médicale; il est donc très inhabituel même dans le cadre de ce syndrome. Le HCMN est également très inhabituel au sein de la population générale; il peut être diagnostiqué à tout âge et la plupart des cas surviennent chez de jeunes enfants de moins de 2 ans. Les personnes atteintes de HCMN présentent une congestion nasale signalant que les voies nasales sont partiellement ou complètement obstruées. Les médecins peuvent déceler la présence de tissus inhabituels dans la cavité nasale. Un tomodensitogramme ou un examen par IRM révèle la présence de tissus mous remplissant la cavité nasale ou les sinus adjacents, ou ces deux endroits à la fois. Il est nécessaire de procéder à une biopsie chirurgicale pour établir le diagnostic.
Comment est le HCMN traité?
L’ablation du HCMN est généralement couronnée de succès. Rarement, plus d’une intervention chirurgicale peut être nécessaire pour éliminer tout HCMN ou les HCMN récurrents (ce qui est inhabituel).
Quand tester pour des mutations dans DICER1?:
Le HCMN, comme le MECC que nous avons abordé ci dessus, est très inhabituel au sein de la population générale et dans le cadre du syndrome DICER1. Selon l’information actuellement disponible, il semble que les personnes présentant une mutation de DICER1 courent un risque légèrement plus élevé d’être atteintes d’un HCMN. Le HCMN se produit également sans lien avec la mutation de DICER. Ainsi, si un enfant reçoit un diagnostic de HCMN, il est raisonnable de s’informer soigneusement pour déterminer si d’autres maladies associées à une mutation de DICER1 ont été décelées au sein de la famille de celui ci. Si c’est le cas, il est très raisonnable de procéder au dépistage de mutations de DICER1 chez l’enfant présentant un HCMN (ou chez les personnes atteintes d’autres maladies). Si aucune autre maladie associée à DICER1 n’est recensée au sein de la famille, il est moins nécessaire de procéder au dépistage de mutations de DICER1, mais il demeure raisonnable de le faire si la famille le souhaite.
Polypes intestinaux juvéniles

Les polypes intestinaux juvéniles sont de petites masses de la taille d’un raisin ou de petites grappes de masses de la taille d’un raisin qui se forment sur la surface intérieure des intestins. Ils peuvent entraîner un blocage partiel de l’intestin et ainsi ralentir le passage du contenu intestinal. Ils sont bénins. Il peut y avoir un seul polype ou y en avoir seulement quelques uns. Ils surviennent au sein de la population générale ainsi que dans le cadre du syndrome DICER1. Ils sont inhabituels dans le cadre du syndrome DICER1 et, bien qu’il n’existe toujours pas de preuves indiquant qu’ils sont directement liés à une mutation de DICER1, il semble exister un lien.
Les polypes intestinaux juvéniles ne sont aucunement liés au syndrome de polypose familiale du côlon (le « gros intestin ») ou du rectum – cette affection porte le nom de « syndrome de polypose adénomateuse familiale » et est associée à une prédisposition excessive au cancer du côlon chez l’adulte.
Comment les polypes intestinaux juvéniles sont-ils diagnostiqués?
Dans le cadre du syndrome DICER1, ces polypes se forment habituellement dans l’intestin grêle et obstruent éventuellement le tractus intestinal, provoquant ainsi des douleurs et un ballonnement. Diverses techniques de radiographie, comme le tomodensitogramme ou un lavement baryté (ou une technique similaire pour les jeunes enfants) permettent de déceler un blocage et peuvent révéler la présence de polypes à l’intérieur du tractus intestinal. Lorsqu’un polype se forme ailleurs dans le tractus intestinal (comme dans l’œsophage ou le rectum, ce qui arrive rarement), d’autres méthodes diagnostiques sont utilisées.
Comment les polypes intestinaux juvéniles sont-ils traités?
On a recours à la chirurgie pour éliminer les polypes et soulager toute occlusion intestinale.
Quand tester pour des mutations dans DICER1?
Les polypes intestinaux juvéniles sont inhabituels dans le cadre du syndrome DICER1 et sont probablement légèrement plus fréquents au sein de la population générale que dans le cadre de ce syndrome. Par conséquent, si un enfant présentait des polypes juvéniles et qu’aucun membre de sa famille n’était atteint d’autres affections liées à DICER1, les médecins auraient raison de ne pas envisager la présence du syndrome DICER1, et il serait inutile de procéder au dépistage de mutations de DICER1. Par contre, si un enfant ayant des polypes était aussi atteint d’autres affections liées à DICER1 ou si des membres de sa famille proche présentaient de telles affections, il serait raisonnable d’effectuer le dépistage de mutations de DICER1.

 Quelques tumeurs inhabituelles du cerveau se forment dans le cadre du syndrome DICER1. Nous abordons plusieurs de celles ci dans une autre section. Une tumeur du cerveau, le blastome de l’hypophyse, est très inhabituelle et est particulièrement caractéristique du syndrome DICER1; nous l’abordons ici.
Quelques tumeurs inhabituelles du cerveau se forment dans le cadre du syndrome DICER1. Nous abordons plusieurs de celles ci dans une autre section. Une tumeur du cerveau, le blastome de l’hypophyse, est très inhabituelle et est particulièrement caractéristique du syndrome DICER1; nous l’abordons ici.
 Une tumeur de l’ovaire (la tumeur à cellules de Sertoli-Leydig) compte parmi les manifestations fréquentes de la mutation de DICER1 chez les femmes. Une autre tumeur de l’ovaire et une tumeur du col de l’utérus (appelée rhabdomyosarcome embryonnaire) sont de très rares manifestations de la mutation de DICER1.
Une tumeur de l’ovaire (la tumeur à cellules de Sertoli-Leydig) compte parmi les manifestations fréquentes de la mutation de DICER1 chez les femmes. Une autre tumeur de l’ovaire et une tumeur du col de l’utérus (appelée rhabdomyosarcome embryonnaire) sont de très rares manifestations de la mutation de DICER1.