
Esta sección es una adaptación del trabajo: “DICER1: mutations, microRNAs and mechanisms” (DICER1: mutaciones, micro RNA y mecanismos), publicado en Nature Reviews Cancer por Foulkes, Priest y Duchaine (PMID 25176334). Para obtener más información, consulte el artículo original
Funciones moleculares
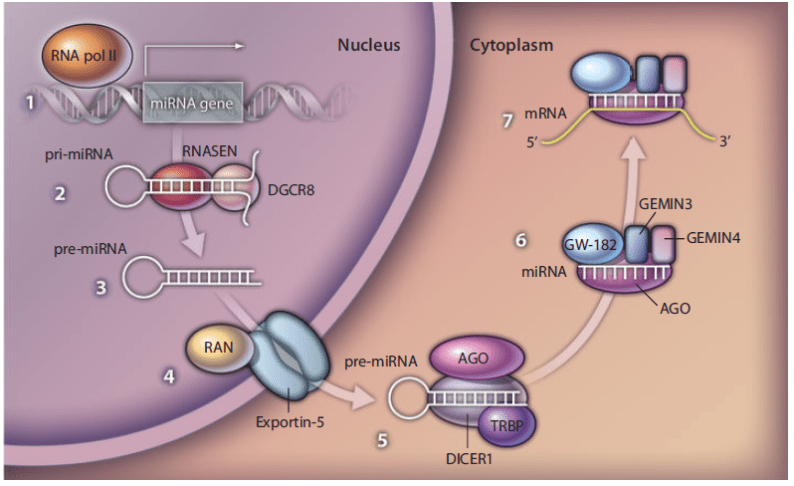
La proteína Dicer es un endoribonucleasa responsable de la producción de micro RNA maduros (miRNA), que son moléculas de RNA de cadena sencilla que tienen como objetivo a un RNA mensajero (mRNA) y poseen un rol importante en la regulación de la expresión génica al reprimir la síntesis de proteínas. La forma humana de la proteína Dicer se denomina DICER1, codificada por el gen DICER1 y se expresa extensamente en varios tejidos. DICER1 participa en la vía de silenciamiento génico mediada por miRNA, donde un miRNA se hibrida de manera imperfecta para abordar los mRNA. A su vez, esto generalmente lleva a una represión de la traducción del mRNA y la iniciación de la degradación del mRNA. Se produce una transcripción del miRNA primario (pri-miRNA) a través de una transcripción de un gen miRNA por la enzima RNA polimerasa II. El pri-miRNA luego es procesado en el núcleo por DROSHA (también conocido como RNASEN), un RNA endonucleasa y la proteína de unión a RNA DGCR8 (también conocido como PASHA), lo cual resulta en la producción de una horquilla de RNA que contiene el miRNA. Esta horquilla de miRNA prematuro (pre-miRNA) se exporta luego al citoplasma a través de los poros nucleares. La proteína DICER1 citoplásmica adhiere la pre-miRNA para liberar el miRNA corto maduro (~21 nucleótidos). Este miRNA luego se carga en una proteína Argonauta (AGO) que forma el núcleo del complejo de silenciamiento inducido por miRNA (miRISC). El miRISC luego se hibrida con los mRNA objetivo para poder generar su silenciamiento.
Estructura de DICER1
- El gen humano DICER1 está ubicado en el cromosoma 14q32.13 y está compuesto por 27 exones con 1,922 aminoácidos. La proteína DICER1 es una enzima grande de multidominio con forma de “L”.
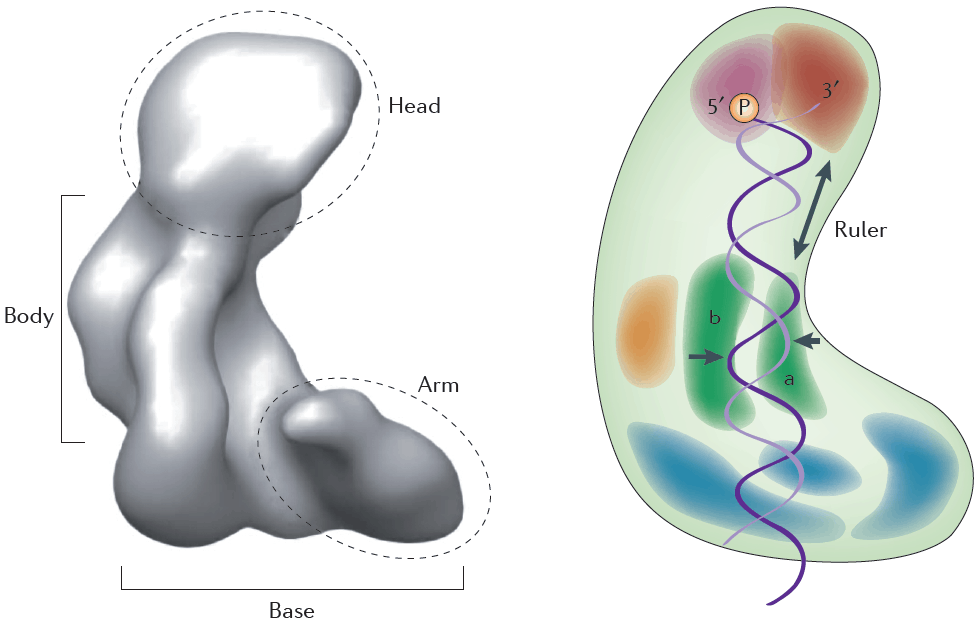
- En la parte superior de la “L” se encuentran los dominios PAZ y Plataforma. Estos dominios son las zonas de enlace para el RNA de doble cadena (dsRNA). La saliente de 3’ del sustrato de dsRNA se une en el dominio PAZ y la saliente de 5’ se une en el dominio Plataforma.
- La parte inferior del DICER1 consta de dominios RNase IIIa y RNase IIIb, que se dimerizan para formar el núcleo catalítico fundamental de la enzima y cada uno adhiere una cadena del sustrato de dsRNA. El dominio RNase IIIa es el responsable de producir 3p miRNA a partir de la cadena 3’ y el dominio RNase IIIb produce 5p miRNA a partir de la cadena 5’. DICER1 requiere iones de magnesio o de manganeso para poder llevar a cabo el clivaje catalítico. Estos iones de metal están unidos a residuos de unión de iones dentro de los dominios RNase III en los residuos de aminoácidos E1320 y E1564 en el dominio RNase IIIa, y D1709 y E1813 en el dominio RNase IIIb.
- Correlación clínica: La mayoría de las personas con tumores relacionados con el síndrome de DICER1 albergan dos mutaciones en DICER1. Generalmente estas consisten de una mutación (inactivante) truncada de línea germinal heterocigótica, que puede estar ubicada en cualquier posición en todo el gen, acoplado a una mutación sin sentido somática (o específica de un tumor), que afecta consistentemente a uno de los residuos de unión de iones de metal dentro del dominio RNase IIIb o un residuo adyacente. Se ha descubierto que casi todos los tumores y displasias asociados al síndrome albergan dichas mutaciones de “zonas de riesgo”. En un subgrupo muy pequeño de pacientes con síndrome de DICER1, los eventos mutacionales iniciales son mutaciones de RNase IIIb en mosaico; el fenotipo en estos casos puede ser más severo.
- Los dominios RNase III están separados de los dominios de unión de dsRNA por un ligador, que contiene una hélice conectora. Esto mantiene el esqueleto de fosfato de dsRNA en su lugar. También actúa como una “regla” al posicionar al pre-miRNA a lo largo de la enzima para que la enzima pueda adherir el dsRNA a los miRNA de los tamaños apropiados.
- En la base de la L se encuentra el dominio DExD/H box helicasa. Se cree que este dominio se sujeta al sustrato dsRNA.
Espectro del tumor del síndrome de DICER1
