Esta seção é uma adaptação do artigo: “DICER1: mutações, microRNAs e mecanismos”, publicado na Nature Reviews Cancer por Foulkes, Priest and Duchaine (PMID 25176334). Para mais informações, consulte o artigo original.
Para obter uma lista atualizada de artigos publicados sobre o gene DICER1 e a síndrome DICER1, clique aqui.
Funções Moleculares
A proteína Dicer é uma endoribonuclease responsável pela produção de microRNAs maduros (miRNAs), que são pequenas moléculas de RNA de fita simples que têm como alvo RNAs mensageiros (mRNAs) e têm um papel importante na regulação da expressão gênica por reprimir a síntese proteica. A forma humana da proteína Dicer é chamada DICER1, codificada pelo gene DICER1, e é amplamente expressa nos tecidos. O DICER1 está envolvido na via de silenciamento gênico mediada por miRNA, onde um miRNA hibridiza imperfeitamente com mRNAs-alvo. Essa hibridização, por sua vez, geralmente leva à repressão da tradução do mRNA e ao início da redução de expressão do mRNA. Um transcrito primário de miRNA (pri-miRNA) é produzido via transcrição de um gene de miRNA pela enzima RNA polimerase II. O pri-miRNA é então processado no núcleo por DROSHA (também conhecido como RNASEN), uma endonuclease de RNA e a proteína de ligação a RNA DGCR8 (também conhecida como PASHA), resultando na produção de um stemloop de RNA que contém o miRNA. Essa estrutura (hairpin ) inicial de miRNA (pre-miRNA) é subsequentemente exportado para o citoplasma através de poros nucleares. A proteína DICER1 citoplasmática então cliva o pré-miRNA para liberar o miRNA curto (~ 21 nucleotídeo) maduro. Este miRNA é então carregado por uma proteína Argonaute (AGO) formando o núcleo do complexo silenciador induzido por miRNA (miRISC). O miRISC hibrida então com mRNAs direcionados para gerar seu silenciamento gênico.
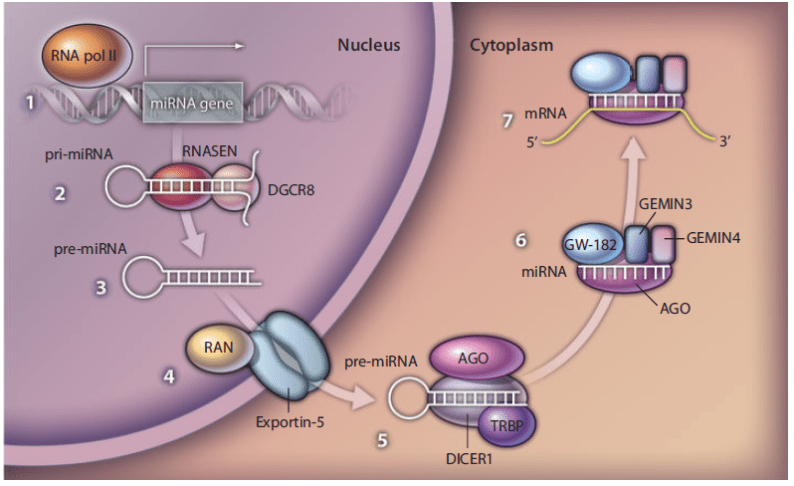
Estrutura de DICER1
O gene DICER1 está localizado no cromossomo 14q32.13 e é composto por 27 exons com 1.922 aminoácidos. A proteína DICER1 é uma grande enzima com vários domínios e em formato de “L”.
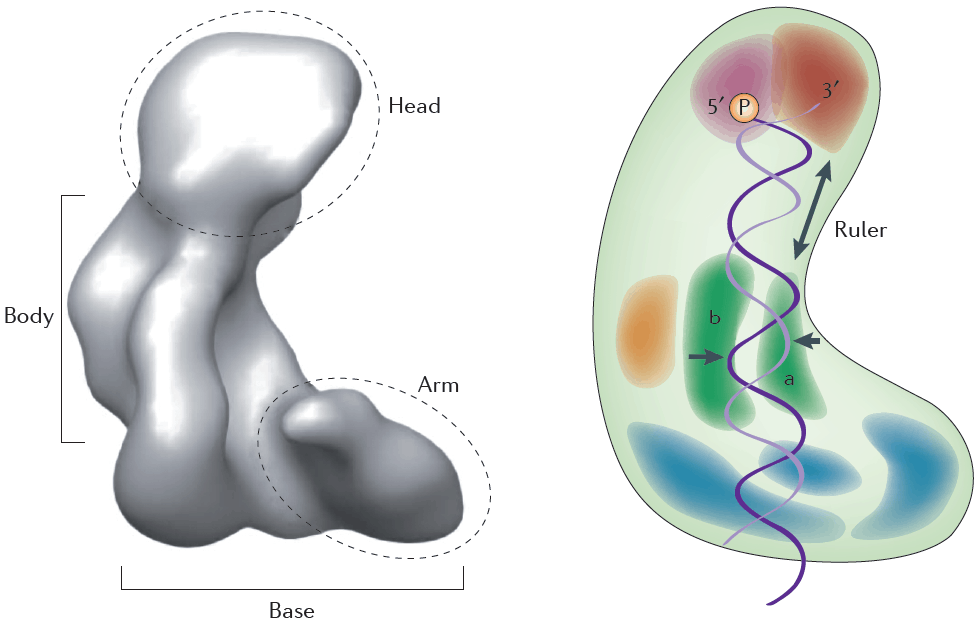
- No topo do “L” estão os domínios PAZ e Platforma. Estes domínios são bolsas de ligação para RNA de cadeia dupla (dsRNA). A protuberância 3 ‘do substrato de dsRNA liga-se ao domínio PAZ e a porção 5’ se liga ao domínio Plataforma
- A metade inferior de DICER1 consiste nos domínios RNase IIIa e RNase IIIb, que dimerizam para formar o núcleo catalítico crítico da enzima, sendo que cada um cliva uma cadeia do substrato de dsRNA. O domínio RNase IIIa é responsável pela produção de 3p miRNAs da cadeia 3 ‘, e o domínio RNase IIIb produz 5p miRNAs da cadeia 5’. O DICER1 requer íons de magnésio e / ou manganês para realizar a clivagem catalítica. Estes íons metálicos estão ligados a resíduos de ligação a íons dentro dos domínios da RNase III nos resíduos de aminoácidos E1320 e E1564 no domínio da RNase IIIa, e D1709 e E1813 no domínio da RNase IIIb.
- Correlação clínica: A maioria dos indivíduos com tumores relacionados com a síndrome DICER1 apresenta duas mutações no DICER1. Estes consistem tipicamente de uma mutação heterozigótica truncada (inativadora) germinativa, que pode estar localizada em qualquer posição em todo o gene, acoplada a uma mutação somática (ou específica do tumor), que afeta consistentemente um dos sítios de ligação de íons metálicos dentro do domínio de RNase IIIb ou um resíduo adjacente. Quase todos os tumores e displasias associados com a síndrome apresentam tais mutações “hotspot”. Em um subgrupo muito pequeno de pacientes com a síndrome DICER1, os eventos mutacionais iniciais são mutações da RNase IIIb em mosaico; o fenótipo nestes casos pode ser mais grave.
- Os domínios da RNase III são separados dos domínios de ligação do dsRNA por um ligante, que contém uma hélice conectora. Isso mantém o backbone de fosfato de dsRNA no lugar. Ele também atua como uma “régua”, posicionando o pré-miRNA ao longo da enzima de forma que a enzima possa clivar o dsRNA em miRNAs de tamanhos apropriados.
- Na base do L está o domínio helicase \ DExD / H. Este domínio é considerado um repressor do substrato de dsRNA.
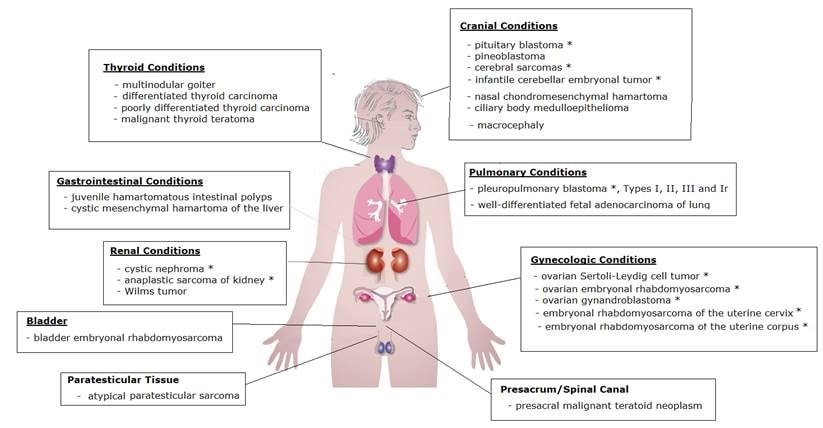
Espectro de Tumores Associados a DICER1